|
|
|
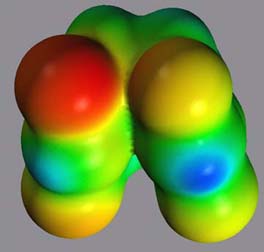
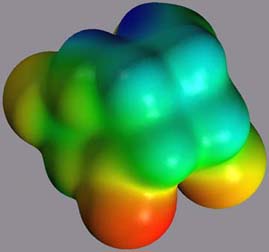
3.3. The cis,syn Cyclobutane Uracil Dimer Radical Anion 1*- As anticipated from the topological differences in the HOMO and LUMO of 1, the geometric and electronic structure of 1*- shows several interesting differences and similarities to 1 and 1*+. As shown in Figure 9, the cyclobutane ring is planarized in 1*-, and the overall structure is almost symmetrical. However, the charge is localized on one of the carbonyl functions in the 4-position, stabilizing the radical anion as a ketyl radical anion. In this conformation, the ketyl radical anion could potentially be stabilized by an interaction of the SOMO with the pi* orbital of the C4' carbonyl. Assuming a binding mode as described above, this charge localization would increase the putative interaction of the C4 carbonyl and a polar group in the ative site and lower the reduction potential of 1. Similar interactions have been described in model systems.[18] The electrostatic potential in the cyclobutyl moiety is very little affected by the reductive electron transfer and the partial charges are almost identical to the ones in neutral 1.
Figure 9: Electrostatic potential of 1*-, projected onto an isodensity surface. left: view of the carbonyl moiety; right: view of the cyclobutane moiety
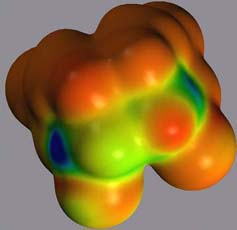
Figure 10 left shows the spin density, projected onto an isodensity surface of 1*-. In agreement with the localized negative charge discussed above, the spin density in 1*- is mostly localized at C4, but extends also towards the C5-C5' bond. Unlike the corresponding C6-C6' bond in 1*+ , this bond is only negligibly stretched since the additional electron resides mostly in the pi* orbital of the carbonyl function.
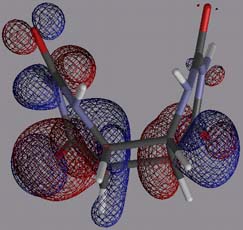
Figure 10 left: Spin density of 1*-, projected onto an isodensity surface; right: SOMO of 1*-
|