|
|
|
The endogenous cannabinoid signalling system: chemistry, biochemistry and physiology.
Vincenzo Di Marzo1 and Luciano De Petrocellis2 1Istituto per la Chimica di Molecole di Interesse Biologico and 2Istituto di Cibernetica, C.N.R.,Via Toiano 6, 80072, Arco Felice, Napoli, Italy Received 28 March 1997 Copyright � 1997 Internet Journal of Science - Biological Chemistry Abstract The cloning of central and peripheral cannabinoid receptor subtypes and the finding, in animal tissues, of endogenous metabolites capable of selectively binding them, opened a new age in cannabinoid research. In this article, starting from the pharmacology and chemistry of Indian hemp-derived as well as synthetic cannabinoids, and ending with the most recent findings on the biochemistry of endocannabinoids, we review the current state of the art of cannabinoid research, and explore the possible physiological roles of a newly discovered chemical regulatory apparatus: the endogenous cannabinoid system. Contents
Pharmacological properties of plant and synthetic cannabinoids Chemical signalling through cannabinoid receptors The discovery of the ‘endocannabinoids’ Metabolic aspects of the ‘endocannabinoids’: biosynthesis, uptake and degradation Possible physiological roles of the endogenous cannabinoid system
Several similarities exist in the centuries-old histories of scientific research on opium and cannabis, the two illicit drugs of plant origin most widely used in the world. In both cases, the isolation and chemical characterization of the plant major active principle allowed its chemical synthesis and, subsequently, the thorough investigation of its pharmacological properties. These studies opened the way to the discovery in mammals of membrane receptors (and of the trans-membrane signal transduction events therewith associated) that could explain these pharmacological actions at the cellular and molecular level. As the presence in animals of receptors for molecules of plant origin could be justified only by the existence of endogenous ligands capable of binding to them, the characterization (1) and subsequent cloning (2) of the first cannabinoid receptor in mammalian brain in 1990 allowed, two years later, the isolation and characterization of the first ‘endocannabinoid’, anandamide (3), much in the same way as the finding of opiate receptors (4) had led to the discovery of ‘endorphins’ in the 1970’s (5). The somehow slower evolution of cannabis research compared to opiate research cannot be explained by a lower interest of the scientific community in cannabinoid pharmacology, since in the early nineteenth century the potentially important therapeutic exploitation of Cannabis sativa preparations, promped by their wide use in Indian folklore medicine, had been already recognized also by several Western physicians (6). It was rather the peculiar chemical nature of cannabis most abundant active principle, the highly lipophilic (-)-D9-tetrahydrocannabinol, that made difficult its isolation and characterization (which were completed only in 1964 [7]), as well as its chemical synthesis and pharmacological handling, thus delaying the discovery of cannabinoid binding sites until 1988 (3). Unlike the ‘endogenous opiate system’, the development of our knowledge on the recently discovered ‘endogenous cannabinoid system’ has greatly benefited from (and is still greatly relying upon) the effort of bio-organic and natural product chemists, whose experience with lipophilic molecules, integrated with the work of pharmacologists, biochemists and molecular biologists, turned out to be extremely useful. In this article, while laying particular emphasis on the role of chemistry in both basic and applied cannabinoid research, we review the milestones that have led to the discovery of the ‘endogenous cannabinoid system’, and discuss the current hypotheses on its possible physiological functions.
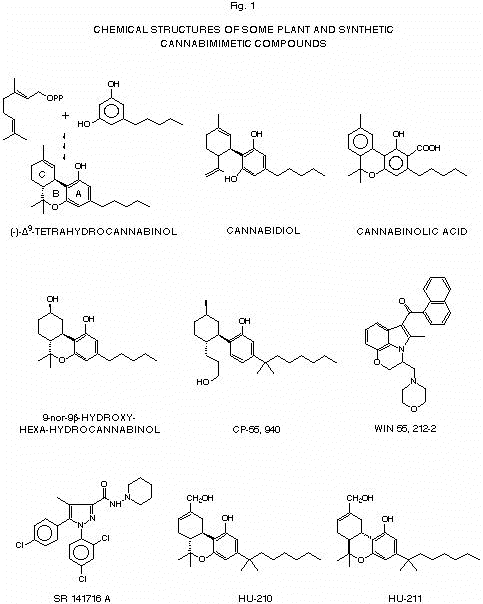
Pharmacological properties of plant and synthetic cannabinoids As it is often the case with recreational drugs, cannabis initial popularity was probably due not only to its pshychoactive properties, but also to its several medical applications, widely documented by Indian medicine, and ranging from treatment of cramps, migraine, convulsions and neuralgia to attenuation of nausea and vomiting, decreased intestinal motility during diarrhea, bronchodilation in asthma and appetite stimulation (6). It was certainly also with the hope of finding potentially useful drugs that the isolation and chemical characterization of cannabis major, and essentially only, psychotropic constituent were painstakingly attempted and finally achieved in 1964 (7) thanks to the efforts of two Israeli natural
The hypothetic precursors for D9-tetrahydrocannabinol biosynthesis in plants, i.e. geranylpyrophosphate and olivetol, are shown. product chemists, Y. Gaoni and R. Mechoulam, who brilliantly completed the pioneering work started in the 1940’s by R. Adams (8). This component, (-)-D9-tetrahydrocannabinol (THC), is one of a family of about 60 bi- and tri-cyclic compunds, formally derived from geranyl-pyrophosphate and olivetol, and named cannabinoids. These natural products usually contain a 1,1’-di-methyl-pyrane ring (the B ring), a variedly derivatized aromatic ring (the C ring), and a variedly unsaturated ciclohexyl ring (the A ring), and include also the non-psychoactive cannabinol, cannabidiol and cannabinolic acid (Fig. 1). The latter compounds have been suggested to contibute to some of THC-mediated effects of cannabis on peripheral tissues, such as cell protection, immunosuppression and anti-inflammatory properties (9). The structure elucidation of THC opened the way to the design of synthetic strategies for this as well as other natural cannabis components and also for a series of analogs (10). By 1986 over 300 such compounds, reviewed by Razdan (11), were available, and an extensive study of the pharmacological properties of THC, previously prevented by the rapid deterioration of crude cannabis preparations, as well as of the structural pre-requisites necessary for this compound to exert its typical psychotropic properties, was finally possible. A long list of pharmacological properties, both in the CNS and in peripheral tissues, was soon compiled for THC (12, 13), including the analgesic, antiemetic, anti-inflammatory, bronchodilatory and anti-convulsant effects already known for cannabis preparations, but also the reduction of blood ocular pressure in glaucomic patients and the alleviation of neurological disorders such as multiple sclerosis, Huntington’s chorea, spinal cord injury-associated spasticity and seizures. Also the illicitly looked for psychotropic properties of marijuana and hashish could be more accurately studied by using synthetic THC, which could reproduce all the typical effects of cannabis in humans (tachycardia, impairment of memory, alteration of mood, motor coordination, posture, cognitive ability and sensory perception) as well as in animal models (dog static ataxia, mouse hypomotility, catalepsy, hypothermy and antinociception) (12, 13). It would be impossible to update the ever increasing inventory of the effects ascribed to THC, which include other beneficial as well as noxious properties such as abortive and anti-fertility actions, various metabolic effects and the modulation of the release and/or action of prostaglandins or of pituitary and steroid hormones (12-14). A thorough structure-activity relationship (SAR) investigation on cannabinoids was originally performed by Martin and coworkers (15) by using a multi-parameter mouse model that included the measurement of: a) antinociception, determined by tail-flick latency, b) catalepsy, by the ring stand test, c) rectal temperature, and d) spontaneous activity in the open-field test. This ‘tetrad’ of behavioural tests was based on four of the several psychotropic properties of THC, that, when evaluated individually, are not selective for any particular class of compounds, but, if assessed together, have ‘proven to be highly predictive of cannabinoids’ (15). Based on the work of Martin and colleagues as well as on previous data (see refs. [16-18] for recent reviews of SAR studies) it was possible to conclude that: 1) the psychoactive effects of THC are enantioselective for the (-)-trans isomers - in the Martin’s ‘tetrad’ of tests the ED50 ranged between 4 and 21 mg/kg for the (-)-enantiomer, while the (+)-enantiomer was inactive at 30 mg/kg (19); 2) the length as well as the lipophilicity of the C3 alkyl chain are extremely important for biological activity - methylation/elongation of the pentyl chain of THC to the 1,1’- or 1,2-di-methyl-heptyl-derivatives, and hydroxylation (the main route for THC metabolism in vivo) respectively increase and reduce its biological activity; 3) the phenolic hydroxy-group is another important pre-requisite for biological activity - its acetylation, glycosilation, sulfation, replacement with an amine, a carboxyl or a methyl group, all resulted in the partial or total loss of cannabimimetic activity (20); 4) the orientation and chemical nature of the C9 substituent is also an important feature of psychoactive cannabinoids - it must protrude into the same face as the phenolic hydroxy-group for the activity to be observed, while its hydroxylation increases biological potency (21, 22); this last pre-requisite, that keeps into account not only the chemical moieties necessary to the cannabinoid pharmacophore but also their three-dimensional conformation, may explain why the natural cannabis component cannabidiol is not active whereas the synthetic 9-nor-9b-hydroxy-hexahydrocannabinol (HHC, Fig. 1) is ten-fold more active than THC.
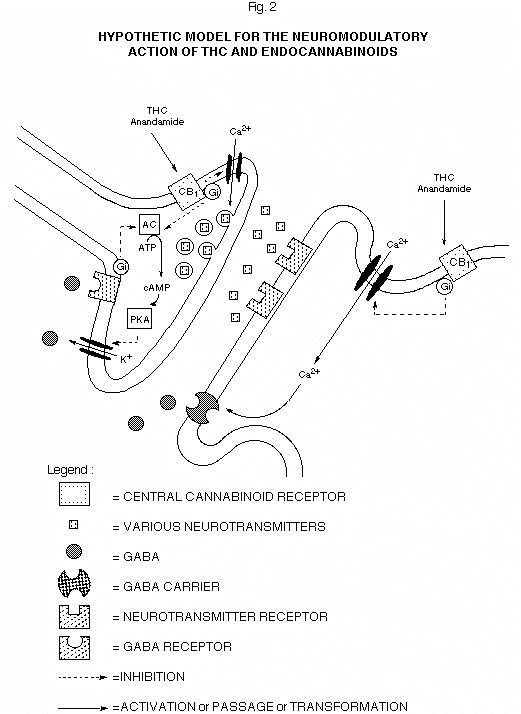
Chemical signalling through cannabinoid receptors Despite the fact that a single mechanism of action is not likely to account for the multiple effects of THC, and that the existence of membrane receptors for this xenobiotic compound has been doubted for several years on the basis of its high lipophilicity, conclusive evidence exists today for the presence in the brain of high affinity and stereoselective cannabinoid binding sites. The stringent structural characteristics that cannabinoid compounds must possess in order to exert their psychological effects was the first strong evidence to a receptor-mediated mechanism of action. Only the existence of proteins with a three-dimensional binding site for cannabinoids could in fact explain, for example, the steric pre-requisites 1) and 4) outlined in the previous chapter. The development of cannabinoid analogs, designed on the basis of these structural requirements and using modern molecular modeling techniques, led eventually to the synthesis of CP-55,940 (Fig. 1), a compound which proved to be 4-25 times more potent than THC and could be radiolabelled and used as a probe for the first characterization of a selective, high affinity cannabinoid binding site (3). This receptor was identified in rat brain cortical membranes, where it exhibited KD and Bmax values respectively of 0.13-5 nM and of 0.9-3.3 pmol/mg protein; its binding with a selected series of cannabinoids perfectly correlated with their antinociceptive potencies (3, 23). The finding of the cannabinoid receptor explained why a completely different class of compounds, the aminoalkylindoles (such as WIN 55,212-2, Fig. 1, [24]), exhibit a range of pharmacological activities in the CNS superimposable to those of THC and other cannabinoids. With the help of computer-assisted molecular modelling it was in fact possible to obtain a three-dimensional model for the cannabinoid binding site whose steric and electrostatic properties kept into account the variations in the potencies of known cannabinoids and accomodated the chemically different aminoalkylindoles (25). The discovery of the ‘central’ cannabinoid receptor accounted for two widely described intra-cellular effects of THC, i.e. the inhibition of agonist-induced cyclic AMP (cAMP) formation and blockade of inward Ca2+ currents mediated by N-type Ca2+ channels (for reviews see [26, 27]). These effects, while suggesting that most of THC actions in the CNS may be due to the interference with neurotransmitters such as g-aminobutyric acid, acetylcholine and the cathecolamines (28), whose action is mediated by cAMP and Ca2+, were shown to be blocked by pre-treatment of cells with pertussis toxin. The latter protein, in turn, catalyzes the ADP-ribosylation and subsequent inactivation of heterotrimeric GTP-binding proteins (the G-proteins) which are coupled to several receptors and deputed to translating an extra-cellular signal into a series of intracellular events. Therefore, it was reasonable to propose that THC, after binding to a receptor on the cell membrane, may activate one of such G-proteins (for example Gi or Go) thereby inhibiting neurotransmitter-induced cAMP formation and Ca2+ influx into neurons, and affecting, as a consequence, other neuronal biochemical events such as neurotransmitter release into and re-uptake from the synaptic interface (Fig. 2).
Figure 2. Anandamide, 2-arachidonoyl-glycerol and psychoactive cannabinoids (e.g. THC) may act at pre-sinaptic CB1 receptors and inhibit, via a pertussis toxin sensitive G-protein, inward calcium currents and adenylate cyclase, thereby influencing, respectively, the release of other neurotransmitters (inhibition) and outward potassium currents (facilitation). The latter effect may lead to potentiation of GABAB receptor-mediated neurotransmission. Endocannabinoids may also act at post-synaptic CB1 receptors and inhibit the re-uptake of neurotransmitters or modulate the likelyhood of action potential generation and impulse propagation'. AC= adenylate cyclase; PKA= protein kinase A; Gi=inhibitory G-protein; GABA=g-aminobutyric acid; THC=D9-tetrahydrocannabinol.
After the first finding in 1988, cannabinoid binding sites were reported by several laboratories in specific brain regions as well as peripheral tissues, such as spleen cells (e.g. lymphocytes and monocytes/macrophages [29, 30]) and reproductive tissues (31), and their distribution was shown to well correlate with THC pharmacological actions. However, final and conclusive evidence for the existence of the central cannabinoid receptor was gained in 1990, when this protein was cloned and sequenced by Matsuda et al. (2) using the ‘homology screening’ approach. Based on the assumption that the protein possessed some sequence homology with other G-protein coupled receptors, an oligonucleotide probe based on the G-protein coupled receptor for substance K was synthesized and used to isolate, from a rat library, a clone encoding for an ‘orphan receptor’, i.e. a receptor with a unique sequence and no known ligand. The receptor was then expressed by transfection of its complementary DNA (cDNA) in host cells and several ligands were screened for their ability to selectively bind to membranes prepared from these cells. Only the cannabinoids were found to bind to these preparations and to inhibit adenylate cyclase-catalyzed cAMP formation. Almost concomitantly to this somehow fortuitous discovery, G�rard et al. reported the isolation and characterization of the human central cannabinoid receptor (32), which displayed a 98% amino acid homology with the rat receptor and was also expressed in the testes (31). The central cannabinoid receptor belongs to the ‘seven trans-membrane spanning receptor’ family, i.e. to those integral membrane proteins whose amino acid sequence assume a three-dimensional conformation with: 1) seven a-helices spanning from one side to the other of the cell membrane, 2) three extra-cellular and three intra-cellular loops, 3) a glycosylated extra-cellular N-terminal domain, and 4) an intra-cellular C-terminal domain involved (together with the intra-cellular loops) in the interaction with the G-protein responsible for the trans-membrane signal transduction of the receptor-mediated signal. In particular, the cannabinoid receptor had 32-39% homology and some structural similarities with the adrenocorticotropic hormone and melanocortin receptors, i.e. the lack of a disulfide bond between the first and second extra-cellular loops and of a proline residue in the fourth and/or fifth trans-membrane domain. The knowledge of the genomic and amino acid
sequence of the central cannabinoid receptor had two important and immediate consequences.
First, it was possible to synthesize oligonucleotide probes for the analysis and
quantitation, assisted also by polymerase chain reaction technology, of the messenger RNA
encoding for the receptor in several tissues (33). Direct visualization of cannabinoid
receptor mRNA could be obtained also by in situ hybridization (34). These studies
confirmed the overall CNS and peripheral distribution of the receptor obtained by previous
binding experiments, even though discrepancies were observed, with some regions containing
little amounts of mRNA and a high density of binding sites and vice versa.
Secondly, three years after the cloning of the central receptor, a second human
cannabinoid receptor could be identified in the marginal zone of the spleen, cloned and
expressed (35). Following to this finding, the central and ‘peripheral’
cannabinoid receptors were named, respectively, CB1 and CB2. Also the CB2 receptor belongs
to the ‘seven trans-membrane spanning receptor’ family, and exhibited an
overall 44% identity with the CB1 receptor (with a 68% identity within the helical
regions), was present in differentiated HL60 cells (35) and in RBL-2H3 basophils (36) but
was not expressed in the brain nor (like the CB1 receptor) in the thymus, liver, lung and
kidneys. Expression, by cDNA The finding of the ‘peripheral’ cannabinoid receptor stimulated new chemical studies aimed at the development of cannabinoid antagonists and agonists selective for either of the two receptors. It was felt that agonists capable of selectively activating the CB2 receptor, being devoid of any psychotropic action, might be used as therapeutically useful compounds. Moreover, the finding of cannabinoid analogs capable of binding selectively only to one of the two cannabinoid receptors, without triggering the activation of the G-protein therewith coupled, might provide pharmacologically and therapeutically useful antagonists. Two landmarks in these new series of experiments were: 1) the synthesis of SR 141716A (Fig. 1), a potent and highly specific CB1 antagonist (43), and 2) the finding that THC binds to the CB2 receptor without inducing a typical cannabinoid receptor-coupled intra-cellular response, i.e. adenylate cyclase inhibition, and behaves as a weak functional antagonist at this receptor (44, 106). More potent, and rather selective, indole ligands for the CB2 receptors have also been described (45, 46), but their functional action on CB2-mediated intracellular events has not been yet assessed. Interestingly, cannabinol, which is not very active at CB1 receptors, was find to bind to CB2 receptors with a relatively low Ki (45). These findings open now the way to studies on the structural features of cannabinoid receptors involved in their interactions, on the one hand, with cannabimimetic ligands, and on the other, with G-proteins (47). A first example of such studies was the identification of lys-192 in the third transmembrane domain of the CB1 receptor as one of the aminoacid residues important for the interaction with HU-210 and CP-55490 but not with WIN 55212-2 (166). Several pieces of evidence seem to suggest the existence of other cannabinoid receptor types both in the CNS and the periphery. Some compounds have been shown to exert typical cannabimimetic actions, such as activation of phospholipase A2, down-regulation of mast cells or inhibition of Ca2+ currents through gap junctions, which cannot be specifically reproduced in cells transfected with either CB1 or CB2 receptors, even though they are blocked by pertussis toxin and/or SR 141716A (37, 54, 104, 114, 119, 153). However, no progress has been made toward the finding of ‘CBn’ receptors, even though an amino-truncated and functionally active isoform of the CB1 receptor has been recently characterized and christened CB1A (48).
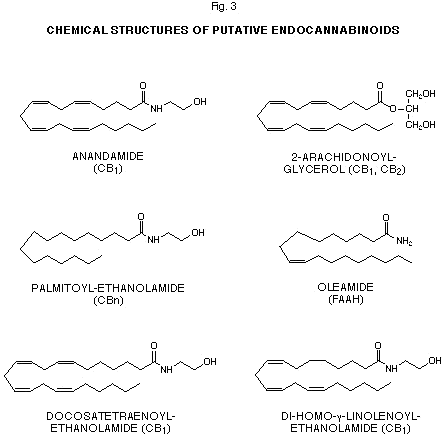
The discovery of the ‘endocannabinoids’ The presence of cannabinoid receptors in mammalian cells could be explained only by assuming the existence of endogenous ligands for these receptors. Studies attempting to test this hypothesis had started already in the late 1980’s but proved to be inconclusive by leading to the isolation of various water soluble brain components whose chemical structure and pharmacological activity could not be fully assessed (49). The idea that an endogenous cannabimimetic metabolite might be, like THC, a lipophilic molecule and not necessarily a peptide, provided a successful lead for the isolation and complete characterization of the first ‘endocannabinoid’. The attention of chemists and biochemists was turned towards the extraction and purification of water insoluble fractions, and it was from such fractions that Devane et al. (3) isolated in 1992 a pig brain constituent capable of binding selectively and with high affinity to CB1 receptor-containing membrane preparations and to inhibit the electrically-stimulated mouse vas deferens twitch response, a typical cannabinoid pharmacological action (50). The chemical structure of this component was elucidated by means of proton NMR and GC/MS analyses. It was immediately clear that the first putative ‘endocannabinoid’ was a derivative of arachidonic acid, a polyunsaturated fatty acid that serves as precursor for a plethora of bioactive metabolites including prostaglandins, thromboxanes, leukotrienes etc. NMR data indicated the presence of amidated ethanolamine too, and MS data confirmed the structure of the metabolite as that of cis-5,8,11,14-eicosatetraenoyl-N-(2-hydroxy-ethyl)-amine (Fig. 3). Due to its possible cannabimimetic psychotropic properties, the compound was named anandamide after the Sanskrit word for ‘bliss’, ananda. During the four years following to its discovery, anandamide was shown to share with THC and other cannabinoids most of their pharmacological properties in both the CNS and peripheral systems, ranging from the basic actions in the ‘tetrad’ of behavioural tests in rodents (51-53), and on the intra-cellular second messengers cAMP and Ca2+ (54-57), to the more peculiar: a) inhibitory effects on memory, motor activity and turning behaviour (58-62), and on ocular blood pressure and heart rate (63, 64), and b) regulatory actions on the levels of hormones of the hypothalamus-pituitary-adrenal axis (65-67), on dopamine-, acetylcholine-, noradrenaline-, endorphin-, glutamate- and GABA-mediated neurotransmission (68-71), and on immune (72, 73) and reproductive (74-76) functions. Cross-tolerance to THC for some of these effects, i.e. inhibition of mouse vas deferens twitch response (77), decrease of motor activity in an open field, catalepsy on a ring, hypothermia, analgesia on a hot plate (78, 79), was also observed with high doses of anandamide, thus further substantiating that this endogenous metabolite acts, at least in part, through the same mechanism as THC. Finally, anandamide, like THC, was found to increase both the affinity and number of rat cerebellum and hippocampus cannabinoid receptors after chronic and acute exposure (80). However, some differences between anandamide and THC actions have been also pointed out. First, the endogenous compound, unlike THC, was suggested to act as a partial agonist for the inhibition of N-type Ca2+ channels in N18 neuroblastoma cells (56). Subsequently, biphasic effects for anandamide were reported by more than a laboratory, with opposite actions with respect to those of THC being observed, both in vivo and in vitro, with very low concentrations of the endocannabinoid (81). The biochemical bases for these effects were not investigated, and it is possible that they be mediated by a high affinity CBn receptor for anandamide coupled to adenylate cyclase through the a subunit of stimulatory (Gs) G-proteins. Another possibility, depicted recently for other Gi-coupled receptors such as the a2-adrenoceptor and the D2-dopamine receptor (82), is that low concentrations of anandamide may still induce the dissociation of pertussis-sensitive inhibitory Gi-proteins with formation of ai subunits in concentrations too low to inhibit type I adenylaye cyclase, and bg subunits capable of activating type II and IV adenylate cyclase in the presence of GTP-bound as subunits. Following to the finding of anandamide in pig brain, novel analytical methods for its purification and quantitative measurement were developed based mainly on HPLC, GC and GC-MS analyses of various derivatives (83-86). Preparation of the 4-(N-chloroformyl-methyl-N-methyl)amino-7-N,N-dimeyhyl-amino-sulphonyl-2,1,3-benzoxadiazole derivatives followed by HPLC and fluorometric detection, provided the first highly sensitive quantitative method (83). Another HPLC method exploited the preparation of the 1-anthroyl-derivatives of anandamide (84). Acetylation of the 2’-hydroxyl group or its derivatization with either trimethyl-silyl-, t-butyl-dimethyl-silyl-, bis-pentafluro-benzoyl- or bis-pentafluoro-propionyl-groups were used for GC and GC/MS methods (85-87). The use of deuterated d8-anandamide (in turn prepared from d8-arachidonic acid) allowed the development of a stable isotope dilution MS technique for anandamide quantitation (87). Anandamide was also directly quantitated without derivatization by using a sensitive LC/MS/MS method (88). By using these methods, anandamide was also identified and quantitated in human, bovine and rat brain (84-89), rat testis (84), rat skin and human and rat spleen (88). Some discrepancies exist in the data, reported by three laboratories (87-89), on the amounts of anandamide in rat brain. SAR studies have been also performed on anandamide in an attempt: a) to explain how a molecule apparently so chemically different from THC could bind to cannabinoid receptors and exert the same actions, b) to design novel drugs potentially useful in therapeutical applications and resistant to enzymatic degradation. It was, in fact, soon clear that anandamide was quickly degraded during both in vivo and in vitro pharmacological tests, as shown by the more rapid onset and termination of its action with respect to THC (51-53), and by the fact that non-specific inhibitors of esterases and proteases such as phenyl-methyl-sulphonyl-fluoride (PMSF) could both prolong and potentiate anandamide effects (90). The affinity for the CB1 receptor was shown to depend on both the length and degree of unsaturation of the fatty acid chain - aliphatic chains with less than three double bonds and with more or less than 20 carbon atoms were less potent or inactive (54). Ethanolamides of w6 polyunsaturated fatty acids were more active than the ones of the corresponding w3 polyunsaturated fatty acids. Hydroxylation of one of the four 1,4-diene groups, obtained by either autoxidation or enzymatic oxidation of anandamide (see also next chapter) and disregarding the stereochemistry of the alcoholic carbon atoms, variedly affected the CB1 binding activity of anandamide, with 12- and 15-hydroxy-anandamides being almost equipotent as anandamide and 8-, 9-, 11- and 5-hydroxy-anandamides being less active or inactive (91-93). Finally, ethanolamides of prostaglandins and leukotrienes were all completely inactive. This suggested that the steric restrictions induced by the formation of prostaglandin-like hairpins or the major mobility and hydrophilicity caused by the introduction of hydroxyl groups disrupt the three-dimensional conformation necessary for anandamide to mimick the THC pharmacophore and correctly orientate its pentyl chain. Modification of the N-(2-hydroxy-ethyl)- moiety of anandamide also proved to cause dramatic effects on the binding to CB1 receptors - its substitution with GABA and L-serine (97), with amides, ethers or esters of various length (94, 95) significantly decreased or abolished the activity, whereas its elongation by one carbon atom increased the potency of one order of magnitude. N-2-(4-hydroxy-phenyl)-ethyl- and N-2-(4-hydroxy-phenyl)-anandamide were less active than anandamide but more resistent to amidase-catalyzed inactivation (96). Finally, substitution of the hydroxyl group with a fluorine atom also increased the activity (95), while the introduction of a methyl group, as in N-(2-hydroxy-isopropyl)-arachidonoyl-ethanolamide (methanandamide), produced a compound less sensitive to enzymativ hydrolysis (98). The ability of this derivative to bind to CB1 receptors depends on the absolute configuration of the new chiral center which has to be R in order to observe an increased activity compared to anandamide (98). These data prompted the formulation of a model for the conformational relationships between anandamide and the cannabinoid pharmacophore (99) which was subsequently ameliorated by computer-assisted molecular dynamics studies (100). The latter showed that a looped, low-energy conformation of anandamide can overlap to the three-dimensional conformation of classical cannabinoids through the superimposition of: 1) the hydroxyl group of the ethanolamide and the phenolic group of THC; 2) the oxygen of the carboxyamide and the pyran oxygen in THC; 3) the w6 aliphatic region of anandamide and the C3 pentyl chain of THC; 4) the polyunsaturated loop and the cannabinoid tricyclic structure. This model can accomodate also other polyunsaturated N-acyl-ethanolamines such as the ethanolamides of di-homo-g-linolenic (20:3, w6) and docosatetraenoic (22:4, w6) acids, which were found in pig brain and, at concentrations slightly higher than those required for anandamide, shown: a) to bind to CB1 receptors (101, 54) and inhibit both forskolin-stimulated adenylate cyclase (54) and mouse vas deferens twitch response (102); and b) to be active in the ‘tetrad’ of mice behavioural tests (103). Recently another study, conducted with over 35 anandamide derivatives, provided further insights into the SAR of the endocannabinoid (104). Surprisingly, the hydroxyl group of the alkanolamine moiety was not found to be essential for CB1 binding activity, as arachidonoyl-n-propylamide and -isopropylamide were both found to be more active than anandamide. On the other hand, a secondary amide group in the molecule was shown to be a necessary pre-requisite for activity. Relevantly to the possible phospholipid origin of anandamide (see next chapter), its phosphorylation abolished binding activity. Finally introduction of one or two methyl groups on the carbon atom a to the carbonyl group in anandamide did not cause any change in the binding activity whereas analogous derivatizations of arachidonoyl-n-propylamide and -isopropyl-amides resulted in the two most active anandamide derivatives so far described (104). Another recent SAR study was carried out with head group analogs of anandamide and (R)-methanandamide, and confirmed the above model (105). More importantly, in this study some of the most active compounds at CB1 receptors, i.e. anandamide, 2’-fluoro-anandamide and (R)-methanandamide, were tested for their binding to CB2 receptors using a mouse spleen preparation and were found to bind very weakly, in agreement with the original finding by Munro et al. (35), and with the suggestion that anandamide, like THC, behaves as a weak agonist at CB2 receptors (106). These findings raise the question of whether there might be other ‘endocannabinoids’ selective for the CB2 cannabinoid receptor and produced in peripheral tissues. Two series of investigations attempted to find an answer to this question and led, respectively, to isolate another cannabimimetic arachidonic acid derivative, 2-arachidonoyl-glycerol, from canine gut (107), and to suggest for a saturated congener of anandamide, palmitoyl-ethanolamide, a role as endogenous CB2 receptor ligand with anti-inflammatory actions on mast cells/basophils (36). 2-Arachidonoyl-glycerol was shown to bind to both CB2 and CB1 receptors, transiently expressed in COS-7 cells, with Ki values higher than those previously reported for anandamide (107). This novel putative endocannabinoid produced in mice the typical ‘tetrad’ of cannabinoid behavioural actions, inhibited forskolin-induced cAMP accumulation (107), and shared with THC a down-regulatory action on mouse splenocyte proliferation (108), while it was much less active than THC and anandamide in inhibiting the mouse vas deferens twitch response (107). Later, 2-arachidonoyl-glycerol was shown to be present also in rat brain, in levels higher than those of anandamide (109), and in dog spleen and pancreas (110). Recently, the cannabimimetic monoglyceride was found to induce a rapid, transient elevation of intracellular Ca2+ concentration in neuroblastoma X glioma hybrid cells, an effect that was also exerted by WIN 55,212-2 and blocked by the CB1 antagonist SR 141716A (167). Although a thorough analysis of its pharmacological properties in both the CNS and peripheral tissues has not been carried out yet, it is possible to hypothesize for 2-arachidonoyl-glycerol a role as either ‘primary’ or ‘auxiliary’ endocannabinoid on those target cells that selectively express either CB2 or CB1 receptors, where anandamide, respectively, has either no significant effect or is the ‘primary’ cannabinoid ligand. The picture is much less clear for palmitoyl-ethanolamide, an N-acyl-ethanolamide which is inactive at the CB1 receptor (54, 55) and is co-synthesized with anandamide in all tissues and cells examined so far (see next chapter). This lipid was shown, together with classical cannabinoids, to potently inhibit serotonin release from rat basophilic leukaemia (RBL-2H3) cells, which express CB2 but not CB1 receptors, and to displace the binding of [3H]WIN 55,212-2 from membrane preparations from the same cells (36). Anandamide was found to antagonize the down-modulatory effects of palmitoyl-ethanolamide and cannabinoids and did not exert any effect on its own. These findings suggested that palmitoyl-ethanolamide and cannabinoid anti-inflammatory effect in RBL-2H3 cells were mediated by CB2 receptors, which cannot be efficiently activated by anandamide (106). However, when palmitoyl-ethanolamide was tested for its binding to membrane preparations from cells selectively transfected with CB2 receptor cDNA, it was found to be inactive (104). This discrepancy can be explained by hypothesizing the presence in RBL-2H3 cell membranes, apart of CB2 receptors, also of a ‘CBn’ receptor different from CB1 and responsible for the immunosuppressive actions of cannabinoids and palmitoyl-ethanolamide, or by assuming for the latter metabolite a non-cannabinoid receptor-mediated mechanism of action. Whatever its mode of action, the mast cell/basophil-mediated anti-inflammatory effects of palmitoyl-ethanolamide, in many aspects similar to those ascribed to synthetic cannabinoids (9), are substantiated by in vivo pharmacological studies conducted both in the 1960-70’s (111) and very recently (112), and by its de novo synthesis by stimulated basophils and macrophages (113). Moreover, a neuroprotective action against glutamate-induced excitotoxicity has been recently described for palmitoyl-ethanolamide in mouse cerebellar granule neurons (114). This effect was again suggested to be mediated by the CB2 receptor (whose mRNA was shown to be expressed in these cells), since it was not exerted, but, on the contrary, was antagonized, by anandamide. Also in this case the effect of palmitoyl-ethanolamide may be due to a non-CB1 non-CB2 receptor, and it is
Figure 3. The proposed target of each metabolite is shown in parentheses. CB1=‘central’ cannabinoid receptor; CB2 ‘peripheral’ cannabinoid receptor; CBn=non-CB1-non-CB2 cannabinoid receptor(s); FAAH=fatty acid amide hydrolase.
noteworthy that neuroprotective actions have been reported for non-psychoactive synthetic cannabinoids such as dexanabinol ((+)-HU-211, Fig. 1) that, like palmitoyl-ethanolamide, do not bind the CB1 receptor, but, unlike palmitoyl-ethanolamide (114), may act as non-competitive antagonists at the N-methyl-D-aspartate receptor for glutamate (115). Apart from 2-arachidonoyl-glycerol and N-acyl-ethanolamines, another potentially cannabimimetic long chain fatty acid derivative, cis-9-octadecenoamide (oleamide), has been recently isolated from the cerebrospinal fluid of cats and humans deprived of sleep and shown to induce sleep in mammals (116). Although not yet demonstrated for anandamide, a sleep-inducing action has been often suggested for the endocannabinoid and THC because of their sedative and motor inhibitory properties, and both slow-wave and REM sleep have been found to be inhibited by the CB1 antagonist SR 141716A (117), suggesting that " .... an endogenous cannabimimetic system may regulate the organization of the sleep-waking cycle". Accordingly, oleamide, like anandamide and/or 2-arachidonoyl-glycerol, was recently shown: a) to be active in the ‘tetrad’ of mice behavioural tests (118) which is highly predictive of cannabinoids, b) to inhibit gap junction-mediated Ca2+ fluxes in astrocytes (119, 120), and c) to inhibit lymphocyte proliferation (121). However, the cannabimimetic actions of this compound cannot be due to the activation of any of the known cannabinoid receptors, since it binds to either CB1 or CB2 receptors in transfected cells only at very high concentrations (> 10 mM, [104, 117, 122]). An alternative, and indirect, way of exerting cannabimimetic actions has been suggested for oleamide (118) based on its ability to inhibit the enzymatic hydrolysis of anandamide (123) and, therefore, to raise the endogenous levels of the latter, by competing for the same inactivating enzyme, i.e. the recently characterized ‘fatty acid amide hydrolase’ (120) (see next chapter). A similar action may be expected also by other long chain fatty acid ethanolamides, which are co-synthesized with anandamide in neurons (124) but do not bind the CB1 receptor (54), and whose biological role is still unclear.
Metabolic aspects of the ‘endocannabinoids’: biosynthesis, uptake and degradation A necessary pre-requisite for an endogenous
metabolite to play a role as a physiological (neuro)modulator in a certain organism is
that molecular mechanisms leading to its de novo biosynthesis and inactivation must
exist in that organism. Since 1992, based on previous knowledge and, again, on the work of
bio-organic chemists, two biosynthetic pathways have been proposed for both anandamide and
2-arachidonoyl-glycerol. The two routes suggested in the 1960’s and 80’s (for a
review, see [111]) for the biosynthesis of saturated, mono- and di-unsaturated fatty acid
ethanolamides, i.e. the direct and energy-free enzymatic condensation between the fatty
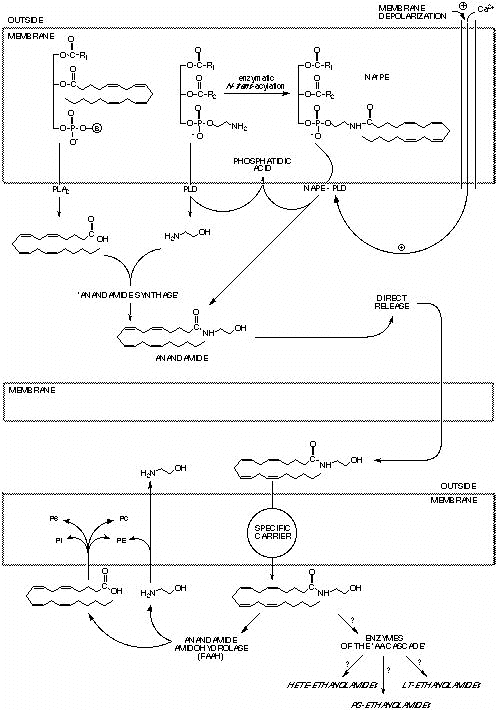
acid and ethanolamine, and the phospholipase D-catalyzed hydrolysis of N-acyl-phosphatidyl-ethanolamines,
have been shown to apply also to anandamide (Fig. 4). The first pathway was found to occur
in mammalian brain particulate fractions independently from ATP and coenzyme A and in the
presence of mM
concentrations of arachidonic acid and high mM concentrations of ethanolamine (126, 127).
The enzyme responsible for this reaction was named ‘anandamide synthase’, was
active at alkaline pH (8.5-10) and selective for arachidonic acid and ethanolamine versus
other fatty acids or amines. As the cellular concentrations of arachidonic acid and
ethanolamine are much lower than those necessary to the enzyme, it was suggested that
‘anandamide synthase’-mediated anandamide biosynthesis would require the prior
activation of the two phospholipases mostly responsible for arachidonic acid and
ethanolamine release from membrane phospholipids, i.e. phospholipase A2
and D. Later, ‘anandamide synthase’ was partially purified from porcine brain
(128) and shown to co-chromatograph with, and to exhibit the same pH/temperature
dependency and inhibition profile as, the enzyme catalyzing anandamide hydrolysis to
arachidonic acid and ethanolamine, ‘anandamide amidohydrolase’ (see below). This
finding, together with the high substrate concentrations required for ‘anandamide
synthase’ to work, allowed to suggest that this enzyme was an ‘anandamide
amidohydrolase’ working ‘in reverse’ (128), and that another pathway,
occurring with more physiologically relevant substrate concentrations, may be responsible
for anandamide biosynthesis in intact cells. This second pathway was shown to underly the
phospholipase A2-independent production of anandamide and other
acyl-ethanolamides in intact rat neurons stimulated with membrane-depolarizing agents
(e.g. ionomycin, kainate and high potassium), and to occur through the
phosphodiesterase-catalyzed hydrolysis of a pre-formed membrane phospholipid precursor, N-arachidonoyl-phosphatidyl-ethanolamine
(N-ArPE) (124). Both N-ArPE and other N-acyl-phosphatidyl-ethanolamines, with the
same N-fatty acid chains as those of the acyl-ethanolamides co-released with
anandamide from stimulated neurons, were characterized in rat neuronal lipid fractions and
their turnover was shown to be induced by ionomycin (124). Moreover, a phospholipase-D
like enzyme catalyzing the conversion of synthetic radioactive N-ArPE to anandamide was
found in rat neuronal homogenates (124), wheras a similar enzyme for the hydrolysis of
other N-acyl-phosphatidyl-ethanolamines to acyl-ethanolamides had been described
and partially characterized from dog brain as early as 1983 (125). Subsequently, these
data were confirmed by another study (89) where acyl-ethanolamides present as lipid
components of rat brain lipid extracts or released by the hydrolysis of the corresponding N-acyl-phosphatidyl-ethanolamines,
purified from the same extracts, were identified and quantitated as 1-anthroyl-derivatives
by HPLC. The presence of a Ca2+-dependent trans-acylase activity catalyzing the
transfer of arachidonic acid from the sn-1 position of 1,2-sn-di-arachidonoyl-phosphatidyl-choline
to phosphatidyl-ethanolamine, with the subsequent formation of N-ArPE (Fig. 4), was also
reported in this study. The enzyme displayed optimal activity at pH=7.0 and Km
values of about 20 and 120 mM, respectively for 1,2-sn-di-arachidonoyl-phosphatidyl-choline
and phosphatidylethanolamine. Following the development of a new HPLC methodology for the
quantitation of N-acyl-phosphatidyl-ethanolamines, the trans-acylase
activity was suggested to be up-regulated, in rat central neurons, by the same stimuli
leading to anandamide and acyl-ethanolamide formation, e.g. augmentation of intracellular
Ca2+ and cAMP concentrations and activation of cAMP-dependent
protein kinase (129). Trans-acylase-catalyzed N-acyl-phosphatidyl-ethanolamine
formation was already known to be induced also by pathological stimuli such as ischemia
and, more generally, tissue injury (111, 130), and anandamide and acyl-ethanolamide levels
in bovine and rat CNS were accordingly shown to be increased post-mortem or
following noxious stimulation (86-88). The phospholipase D-mediated pathway was also
implicated in anandamide and palmitoyl-ethanolamide formation in N18 neuroblastoma cells
(131) and in two immortalized immune cell types, stimulated with either ionomycin or
immunological challenge (113), as well as in sea urchin ovaries (132) and rat testes (84).
Therefore, several pieces of evidence support the involvement in physiopathological
situations of this second pathway of anandamide biosynthesis, which may explain also the
formation of other potential cannabimimetic acyl-ethanolamides (101) in both the CNS and
peripheral tissues. However, the recent finding in the mouse uterus (133) of two distinct
enzymatic activities for the synthesis and degradation of anandamide, respectively from
and to arachidonic acid and ethanolamine, has re-opened the question as to which of the
two proposed pathways is Figure 4. Possible biosynthetic and catabolic pathways for anandamide and related acyl ethanolamides. Anandamide (and palmitoyl-ethanolamide) produced by either a ‘synthase’ enzyme or N-acyl-phosphatidyl-ethanolamine-specific phospholipase D (NAPE-PLD) following membrane depolarization is released outside the cell and acts at neighboring cells. In order to be catalyzed by the ’synthase’ enzyme the formation of anandamide must be preceeded by phospholipase A2 (PLA2) activation. Once released by cells, anandamide or palmitoyl-ethanolamide can be uptaken by selective carrier mechanisms and degraded by anandamide amidohydrolase (fatty acid amide hydrolase=FAAH), thereby producing ethanolamine and fatty acids. The latter are readily incorporated into membrane phospholipids (PC, PS, PE, PC). Alternative catabolic pathways for anandamide may utilize the enzymes of the arachidonic acid (AA) cascade; however the formation of the ethanolamides of hydroxy-eicosatetraenoic acids (HETE), prostaglandins (PG) or leukotrienes (LT) has never been in intact cells. B= phospholipid bases.
actually responsible for the biosynthesis of the endocannabinoid in vivo (for a recent review see also [134]). As with anandamide, also the biosynthesis of 2-arachidonoyl-glycerol was shown to be stimulated by ionomycin in neuronal cells (135). In mouse neuroblastoma cells, this effect was Ca2+-dependent, accompanied by 1-acyl-2-arachidonoyl-glycerol formation, potentiated by incubation of cells with exogenous phospholipase A2 , and not sensitive to an inhibitor of one of the enzymes responsible for 1-acyl-2-arachidonoyl-glycerol formation in cells, i.e. phospholipase C. An abundant enzymatic activity catalyzing 1-acyl-2-arachidonoyl-glycerol hydrolysis to 2-arachidonoyl-glycerol was also found in neuroblastoma cell homogenates (135) together with phospholipase C-like enzymes capable of slowly but significantly catalyzing the conversion of sn-2-arachidonoyl-containing (lyso)phosphatidylcholine species into 1-acyl-2-arachidonoyl-glycerol (136). It was suggested that 2-arachidonoyl-glycerol biosynthesis may follow either phospholipase A2- or phospholipase C-mediated pathways depending on whether the neuron is stimulated with agents that are coupled only to Ca2+ influx or also to phospholipase C
Fig. 5
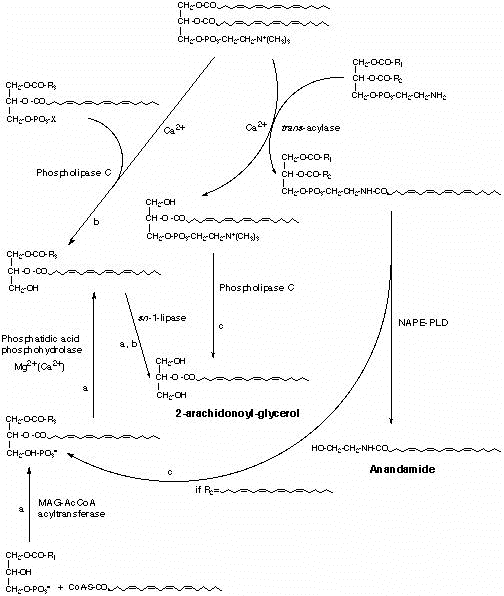
Fig. 5. Potential biosynthetic connections between anandamide and 2-arachidonyl glycerol in mouse neuroblastoma cells. a= Phospholipase A2-dependent pathway; b= pathway in competition with anandamide biosynthesis; c= pathways occurring concomitantly to anandamide biosynthesis. MAG-AcCOA=monoacylglycerol-3-phosphate:acyl-coenzymeA; 2-AG= 2-arachidonoylglycerol; NAPE-PLD= Phospholipase D selective for N-acyl-phosphatidyl-ethanolamines.
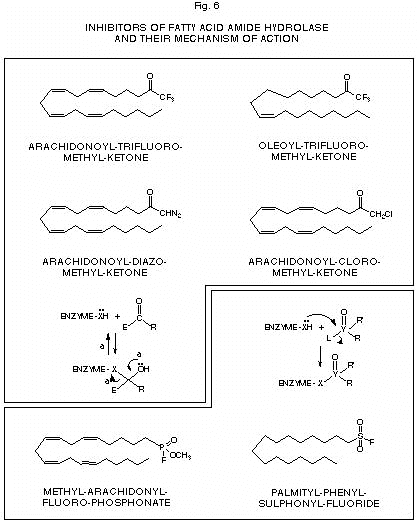
activation. The participation of phosphatidic acid, produced either from de novo biosynthesis or by phospholipase D-activation, as precursor for 1-acyl-2-arachidonoyl-glycerol may be also hypothesized, but still needs to be investigated. It is also possible that, under certain circumstances, the de novo formation of 2-arachidonoyl-glycerol may occur concomitantly to that of acyl-ethanolamides, including anandamide, starting from sn-2-arachidonate-containing 1-lyso-phosphatidylcholine (136) or phosphatidic acid, two by-products of acyl-ethanolamide biosynthesis (Fig. 5). A preliminary study has recently cast some light also on the biosynthesis of oleamide in rat brain. An enzymatic activity catalyzing the formation of the cannabimimetic sleep-inducing factor from oleic acid and ammonia was in fact identified in rat brain microsomes (168), and suggested to be the same as anandamide synthase-amidohydrolase. Metabolic pathways, leading to partial or complete inactivation of anandamide and 2-arachidonoyl-glycerol, have also been described. For the former compound, enzymatic hydrolysis of the amide bond is the major degradative pathway (111, 124, 125). This reaction is catalyzed by an enzyme originally named ‘anandamide amidohydrolase’, characterized from rat, mouse and porcine brain particulate fractions, and shown to be optimally active at alkaline pH (8.5-10), quite selective for anandamide versus other acyl-ethanolamides, and inhibited by typical serine- and cysteine-protease inhibitors such as PMSF, p-hydroxy-mercuri-benzoate, p-bromo-phenacyl-bromide and N-ethyl-maleimide (123, 128, 137-140), as well as by the phospholipase A2 inhibitor arachidonoyl-trifluoro-methyl-ketone (141). Interestingly, this enzyme displayed a tissue distribution and a pH dependency profile almost identical to those of an amidohydrolase previously reported to catalyze the hydrolysis of oleoyl-ethanolamide in dog brain (125). Initial efforts toward the full purification of this integral membrane protein proved to be inconclusive. It was only at the end of 1996 that, once again thanks to the contribution of organic chemists, it was possible to fully purify the enzyme. Cravatt et al. were working on the enzyme ‘oleamide hydrolase’ that catalyzes the hydrolysis of the amide bond of oleamide (116, 142). This enzyme had been previously partially characterized first from mouse neuroblastoma N18 cells and subsequently from rat liver, and in both cases suggested to be the same protein as ‘anandamide amidohydrolase’ (123, 143). A chromatographic resin was derivatized using oleoyl-trifluoro-methyl-ketone, a compound previously shown (143) to inhibit ‘oleamide hydrolase’ by forming a reversible, albeit relatively stable, hemiketal adduct with the catalytically active sulphidryl or hydroxyl group (Fig. 6). The derivatized resin was then used for the purification of the enzyme, solubilized from rat liver membranes, by affinity chromatography, which led to the protein pure to homogeneity in basically one single step (120). Once purified the enzyme, it was then not difficult to obtain some amino acid sequence necessary to synthesize oligonucleotide probes or to raise antibodies for the screening of rat DNA libraries and the isolation of the clone containing the gene encoding for the enzyme. The gene was then sequenced and yielded the complete amino acid sequence of the protein, whose cDNA was expressed in COS-7 cells. It was thus possible to confirm the molecular weight of ‘oleamide hydrolase’ (60-65 kDa) and the fact that the enzyme recognizes anandamide as the preferential substrate. This prompted the authors the use of the name ‘fatty acid amide hydrolase’ (FAAH) for this enzyme (120), as previously suggested also for mouse neuroblastoma anandamide amidohydrolase (123). The sequence analysis of FAAH showed a) a hydrophobic region predicted to assume a trans-membrane a-helical conformation, b) a significant homology with other amide hydrolase enzymes, and c) a consensus class II-SH3-domain binding sequence, which suggested that other proteins may interact with FAAH, thereby regulating its activity and/or subcellular localization. Southern and Northern blots of DNA and mRNA from various rat tissues, probed with an internal 800 base pair fragment of FAAH cDNA, confirmed previous data (138) on the tissue distribution of this protein, whose levels are highest in the liver, brain and kidneys, measurable in testes and lung, low in the spleen and undetectable in the skeletal muscle and heart (120). FAAH-like enzymes have been recently described also in rat cortical astrocytes (124) as well as in other peripheral tissues such as sea urchin ovaries (132), mouse uterus (133), ocular tissues (144) and RBL-2H3 basophils (113). In the latter cell type, FAAH, unlike the enzyme from brain and neuronal cells, displayed a high affinity also toward palmitoyl-ethanolamide, in support of the putative role suggested for this compound as endocannabinoid down-regulator of basophils/mast cells. By using several long chain acyl-ethanolamides as substrates, it was shown that the basophilic enzyme had the higher catalytic efficiency the shorter and the more unsaturated the substrate fatty acid chain (113), with the highest efficiency being observed with anandamide, linoleoyl-ethanolamide and palmitoyl-ethanolamide. The possibility of RBL-2H3 cells expressing an isoform of FAAH structurally different from that characterized in brain and rat liver particulate fractions is currently under investigation. Chemical studies have been performed with FAAH. Recently, inhibitors with different mechanisms of action have been obtained from the derivatization of arachidonic acid and/or oleic acid with functional groups potentially reactive towards catalytically active serine and cysteine residues (Fig. 6). Diazo-methyl- and chloro-methyl-derivatives (143, 145) were found
Upper panel: reversible inhibitors forming tetrahedral hemiketal intermediates with catalytically active serine or cysteine groups (a shows the reversal reaction). Lower panel: irreversible, covalent inhibitors forming stable adducts with catalytically active serine or cysteine groups. X= sulphur(II) or oxygen atom; E= strongly electrophilic group; Y=phosphorus or sulphur (VI) atom; L= leaving group.
to potently inhibit FAAH from different sources (for the arachidonoyl-analogs, IC50 values ranged between 2 and 23 mM, [145]), possibly by forming reversible tetrahedral adducts with the active site -SH or -OH groups. The methyl-arachidonoyl-fluoro-phosphonate derivative was the most potent FAAH inhibitor so far described, with IC50 values between 1 and 3 nM, and formed a covalent, enzymatically inactive adduct with the enzyme (145). Palmityl-sulphonyl-fluoride, although less active than arachidonoyl-methyl-fluoro-phosphonate, is another very potent inhibitor of anandamide enzymatic hydrolysis (IC50 =50 nM, [146]) and is also likely to behave as irreversible inhibitor of FAAH, since its analog PMSF has been shown to covalently inhibit the enzyme (145). Data on the mechanism of action of FAAH inhibitors may provide important informations on the sterical and chemical requirements of the enzyme active site as well as on the possible use of these substances for the affinity-labelling of FAAH enzymes. Apart from amide bond hydrolysis, anandamide has been shown to undergo, in tissue homogenates, also to another series of metabolic reactions consisting of the oxidation of its double bonds with subsequent formation of hydroperoxy- and hydroxy-anandamide derivatives. These reactions may be catalyzed by cytochrome p450 hydroxylases (92), even though anandamide has been shown to be recognized as a substrate also by lipoxygenase enzymes (91, 93). Indeed, it is quite possible that anandamide may be recognized also by other enzymes involved in arachidonic acid and polyunsaturated fatty acid metabolism. For example, the endocannabinoid has been shown to be a good substrate for an unusual algal enzyme, an ‘arachidonic acid conjugated-triene isomerase’ (147), with formation of the conjugated 5Z, 7E, 9E-triene derivative, a compound which was found to retain significant CB1 receptor binding activity (148). This finding opens the possibility that anandamide derivatives may be obtained in the future also by enzymatic methods, thus widening the range of compounds to be tested for cannabinoid activity and to be used for SAR studies. If anandamide, palmitoyl-ethanolamide and oleamide are mostly inactivated through the action of the same or highly homologous FAAH enzymes, the breakdown of 2-arachidonoyl-glycerol to arachidonic acid and glycerol was found to be catalyzed by a different type of enzyme(s), located in both particulate and cytosolic fractions of mouse neuroblastoma cells (135). In fact, the enzymatic hydrolysis of the mono-glyceride was not inhibited by either anandamide or arachidonoyl-trifluoro-methyl-ketone, thus ruling out the involvement of FAAH-like enzymes. It is possible that one or more of the several mono-, di- and tri-acylglycerol lipases present in animal cells contribute to 2-arachidonoyl-glycerol degradation, and, unlike anandamide, there is no evidence to date for the existence of a specific enzyme deputed to the physiological inactivation of the cannabimimetic glyceride. Preliminary results, however, have shown that, much in the same way as anandamide enzymatic degradation is inhibited by other acyl-ethanolamides, unsaturated mono-acyl-glycerols may counteract the degradation of 2-arachidonoyl-glycerol and enhance its cannabimimetic activity (149). Finally, uptake mechanisms for cannabimimetic acyl-ethanolamides have been also reported in cultured rat central neurons and cerebellar granule cells (124, 150) as well as RBL-2H3 cells and J774 mouse macrophages (113). These mechanisms are likely to be mediated by specific carrier proteins in asmuch they are: 1) saturable - with Km and Vmax values ranging between 25 and 41 mM and 0.4 and 0.6 nmols min-1, respectively, 2) temperature dependent - most of the uptake is abolished by lowering the temperature to 4�C, 3) selective for anandamide in the CNS and for either anandamide or palmitoyl-ethanolamide in leukocytes, and 4) inhibited by phloretin and alkylating agents such as PMSF or N-ethyl-maleimide. The purification and further characterization of these proteins have not yet been attempted.
Possible physiological roles of the endogenous cannabinoid system Despite the several studies carried out on the pharmacological activity of both plant and endogenous cannabinoids, the physiological role of the endogenous cannabinoid system has remained elusive. While the picture of cannabinoid receptor distribution is becoming more and more clear, still very little is known on the correlation of certain physiopathological responses with the production and inactivation of endocannabinoids and the expression/activity of the enzymes involved. The development of GC/MS techniques for the measurement of anandamide renders now feasible studies on the occurrence of altered levels of this endocannabinoid during certain disfunctions of both central and peripheral systems. The preparation of antibodies against anandamide, and the subsequent setting up of selective radioimmunoassays for this acyl-ethanolamide, is likely to provide, in the future, a more accessible technique for its quantitative determination, and, therefore, to stimulate further studies in this direction. The recent cloning and sequencing of FAAH, i.e. the enzyme most likely responsible for the inactivation of cannabimimetic fatty acid amides, will allow the assessment of its expression in several tissues under physiopathological conditions, which, like with other inactivating enzymes for (neuro)transmitters and (neuro)modulators, will help clarifying the physiological roles of these metabolites. Conversely, the enzymes responsible for acyl-ethanolamide biosynthesis have not been characterized yet, and little information is available on their regulation. From the limited amount of data acquired so far, the general impression is that the endogenous cannabimimetic system may be a widespread tuning system of numerous finely regulated tasks performed in multi-cellular organisms, and that its importance is not limited to central nervous functions but, on the contrary, may concern with several peripheral processes such as the modulation of neurotransmitter release/action at autonomic and sensory fibers, and the control of immunological, gastrointestinal, reproductive and cardiovascular performance. In the brain, this regulatory system, through the action at CB1-like receptors of anandamide, polyunsaturated acyl-ethanolamides and 2-arachidonoyl-glycerol, may interact with thermoregulatory centres as well as regulate perceptive (hearing, colour vision, taste), cognitive (sleep, long term potentiation, short-term memory) and motor (movement, coordination, posture, skeletal muscle tone) functions. Indeed, the presence of CB1 receptors and/or anandamide and/or FAAH in the thalamus, hippocampus and cortex or in the striatum, substantia nigra and cerebellum, respectively, supports a role of the endogenous cannabinoid system in cognitive and motor responses. The recently reported CB1-mediated activation by anandamide of neuronal focal adhesion kinase in hyppocampal neurons (151), has suggested for the endocannabinoid a role in synaptic plasticity. A regulatory action on astrocyte Ca2+ homeostasis through inhibition of gap junctions has been also observed for anandamide and oleamide (119, 120). A role in brain development has also been suggested (169). The physiological relevance to these central functions of the recently reported effect by anandamide on protein kinase C in vitro (170) remains to be assessed. A neuroprotective role of acyl-ethanolamides in general and of palmitoyl-ethanolamide in particular have also been described also on the basis of their production at sites of neuronal damage or death (86-88, 111, 125). Finally, the presence of CB1 receptors in the hypothalamus may imply a role of the endogenous cannabinoid system in the fine tuning of the secretion of pituitary hormones (65-67). In particular, the activation of the release of the hypothalamic hormone causing the secretion of adrenocorticotropic hormone from the pituitary, i.e. corticotropin releasing factor-41, and the subsequent enhancement of corticosterone production from the adrenal gland, have been observed following intracerebroventricular injection of anandamide (65). Likewise, the release of other hormones, such as prolactin and the luteinizing, follicle stimulating and growth hormones may be inhibited by anandamide acting at the level of the hypothalamus (66, 67). The central role of the endogenous cannabinoid system may be effected through an action at both the presynaptic and post-synaptic level. In the first case, endocannabinoids, by acting at presynaptic cannabinoid receptors, and subsequently modifying the intracellular levels of cAMP, K+ and Ca2+ and/or the activity of protein kinases, may modulate the action of other presynaptic neurotransmitters or the release of mediators acting at post-synaptic receptors. Recently a facilitatory action of THC on the release of dynorphins acting at either k- or d-opioid receptors has been described (152), and this mechanism may underly the well known analgesic actions of THC and anandamide. Post-synaptic cannabinoid receptors may mediate an endocannabinoid action on the uptake of neurotransmitters such as GABA, thereby potentiating their action (Fig. 2). Interactions with other neurotransmitters, such as dopamine, acetylcholine and glutamate, may be mediated by similar pre- and post-synaptic actions of endocannabinoids (68-71). In the periphery, the endogenous cannabinoid system may mediate the chemical communications between different types of immune cells or between sensory fibers and blood cells. In acute inflammatory reactions, basophils and mast cells, once activated by IgE receptor cross-linking and/or Ca2+ influx, would release palmitoyl-ethanolamide and anandamide (113) together with histamine, serotonin and leukotrienes. While the latter mediators produce vascular permeability and chemotaxis, palmitoyl-ethanolamide would act as an autacoid signal on the same or on neighboring basophils and mast cells and inhibit their degranulation (36), thus keeping the inflammatory reaction under control. Anandamide might play a dual role by potentiating the activity of basophil/mast cell phospholipase A2 (153), thus facilitating the formation of eicosanoids with often opposing actions on immune cell functionality, e.g. leukotrienes and prostaglandin E2, which respectively induce the recruitment of neutrophils and macrophages and suppress the activity/proliferation of lymphocytes. Basophilic anandamide might enhance the production of prostaglandin E2 from macrophages (where this eicosanoid is one of the major products of AA oxidation) or directly inhibit the proliferation of lymphocytes recruited in the late phase of the inflammatory reaction, while inducing their apoptosis (73). Palmitoyl-ethanolamide and anandamide produced by macrophages might: a) feed-back on macrophages (72), basophils and mast cells (36), again in order to prevent the excessive propagation of the inflammatory response and inhibit the subsequent delayed hypersensitivity (111, 112); b) co-adjuvate macrophage prostaglandin E2 in the non-specific immunosuppression of T helper cells (73), thus participating in the down-regulation of autoimmune reactions. 2-arachidonoyl-glycerol might act as a natural substitute for anandamide on target cells which express CB2 but not CB1 receptors. Were biosynthetic mechanisms similar to those described previously for central neurons (124) to be found for acyl-ethanolamides and 2-arachidonoyl-glycerol also in sensory neurons of afferent C-fibers, these compounds might play a major role in the model of neurogenic inflammation proposed in the 1980’s (154), by acting, together with somatostatin and vasoactive intestinal polypeptide, as ultimate terminators of inflammatory responses generated by noxious stimuli through an ‘axon-reflex’. On the other hand, the finding of presynaptic cannabinoid receptors on sensory neurons, whereupon palmitoyl-ethanolamide and anandamide produced by stimulated leukocytes may act by modulating the release of inflammatory neuropeptides, would add further complexity to this scenario and widen the range of possibilities for feed-back signalling from immune cells to peripheral fibers. The recent report of AnNH-induced inhibition of capsaicin-evoked substance P and CGRP release from the dorsal horn of rat spinal cord (155) provides preliminary support to this hypothesis. Auxiliary leukocytes (basophils and mast cells) and macrophage-like cells, either recruited from local blood vessels upon request or as resident populations, are found in several tissues where they participate in tissue damage-induced immune defense. Therefore, the finding that some of these cells synthesize cannabimimetic acyl-ethyanolamides (113) opens the way to a novel mode for their chemical signalling with several tissues and cell types expressing cannabinoid receptors. For example, the cellular source of acyl-ethanolamides with saturated or monounsaturated N-acyl chains and of their phospholipid precursors in infarcted dog heart and ischemic brain (125) has never been investigated. It is possible that leukocytes recruited from blood following tissue damage may be responsible for the production of acyl-ethanolamides with vasodilatory and depressor activity (64) or neuroprotecting effects (114). If shown to share with macrophages and RBL-2H3 cells the capability of biosynthesizing and responding to acyl-ethanolamides, populations of macrophages (as microglia) and mast cells resident in the brain may establish a network of chemical communications also with central neurons, which contain cannabinoid receptors and synthesize acyl-ethanolamides during physiopathological responses. Finally, endocannabinoids may also act by modulating nitric oxide (NO) release from leukocytes. A strong enhancement of human monocyte and mussel microglia and immunocyte basal NO production, that could be blocked by SR 141716A, has been recently reported for anandamide and CP 55,940 (156); conversely, THC, by decreasing cAMP levels, was found to inhibit inducible NO synthase expression and NO production by a macrophage cell line that does not express CB1 receptors (157). Therefore, different responses seem to be observed depending on whether constitutive or induced NO formation is measured. It is also possible that, as for other immunomodulatory effects of cannabinoids, the action on NO release from immune cells may depend on which cannabinoid receptor is expressed in these cells. To date, it is not yet clear which of the immune actions of endocannabinoids are mediated by CB1, CB2 or CBn cannabinoid receptors, and a thorough examination of the types of cannabinoid receptors expressed by the different sub-populations of each immune cell type must be carried out in order to obtain a complete picture of the immunomodulatory role of the endogenous cannabinoid system. Mechanisms analogous to the ones described above have been proposed to occur during a hypothetic modulation by the endogenous cannabimimetic system of cardiovascular, respiratory, digestive and escretory functions, particularly for what concerns with the control of smooth muscle tone. Endocannabinoids and THC were shown to inhibit guinea-pig small intestine and mouse urinary bladder acetylcholine-induced contractions by a pre-synaptic, CB1 receptor-mediated inhibitory action on the release of acetylcholine from autonomic fibres (158, 159). The hypothensive effects of anandamide and THC were also shown to be due in part to a presynaptic action on CB1 receptors and to the subsequent inhibition of noradrenaline release from peripheral sympathetic nerve terminals innervating the heart and vasculature (160, 161). Very recently, Randall and colleagues reported intriguing observations suggesting that the endothelium-derived hyperpolarizing factor (EDHF), whose structure has remained elusive since its existence was suggested some years ago, may be anandamide. EDHF-mediated relaxations in the rat mesenteric arterial bed were blocked by SR 141716A, and were accompanied by the formation of an arachidonate metabolite co-eluting with anandamide on TLC. Moreover, synthetic anandamide was found to relax the mesentery through an hyperpolarizing mechanism (171). The physiological function of the endogenous cannabinoid system is not necessarily mediated by either central or peripheral neurons. Apart from immune cells, also tissues of the reproductive system have been shown to both contain cannabinoid receptors and synthesize as well as degrade putative endocannabinoids such as the cannabimimetic acyl-ethanolamides, which may act as local autacoid mediators. Anandamide and/or CB1 receptors have been found in rat testes and mouse vas deferens (32, 84, 162), thus allowing to hypothesize a role of the ‘anandamidergic’ system in the control of spermatogenesis and male fertility. Cannabinoid receptors have been found also on sea urchin sperm cell membrane and suggested to mediate THC inhibition of the acrosome reaction (163, 164). Anandamide has been shown to inhibit the acrosome reaction (74) and sea urchin ovaries have been found to synthesize and degrade anandamide and palmitoyl-ethanolamide (132). It was proposed that sea urchin eggs may synthesize anandamide during the acrosome reaction with the purpose of inhibiting polyspermic fertilization. It remains to be assessed whether an analogous mechanism also occurs in mammals. Both CB1 and CB2 receptors have been found in mouse uterus, which contain also two distinct ‘anandamide synthase’ and FAAH-like enzymatic activities, whose levels appear to vary within the embryo implantation and inter-implantation sites (75, 76, 133). Apart from a possible metabolic action on uterin lactoferrin production (75), the endogenous cannabinoid system was suggested to mediate the chemical communication between the uterus and the embryo, as indicated by the presence of CB1 and CB2 receptors also in embryos starting from very early stages of their development (from the one-cell to the blastocyst stages [76]). Due to its inhibitory effect on embryonic cell division (76), anandamide may act as a negative signal for embryo development and implantation, as shown also by the higher and lower levels, respectively, of anandamide synthase and amide hydrolase detected when the uterus is least receptive to implantation (133). Therefore, the synthesis of anandamide may be used by the uterus in order to direct the timing and placing of embryo implantation. After birth, behavioural responses to anandamide develop gradually until adulthood, as shown by recent ontogenetic studies carried out in young mice. This suggests that, although being present since the early stages of development, CB1 cannabinoid receptors do not reach immediately their full development in the brain (165).
As suggested by a recent article, the discoveries of the first endocannabinoid, anandamide, in 1992, and of peripheral cannabinoid receptors in 1993, have represented ‘a new dawn for cannabinoid research’ (178), by substantiating, respectively, the existence of a new endogenous regulatory system and the expectancy of new drugs, devoid of psychotropic actions, from cannabimimetic compounds. However, the general feeling among those working on this subject that the present knowledge of the endogenous cannabinoid system may be only the ‘tip of the iceberg’ allows to foresee that the potential of these discoveries may be even higher than that initially predicted. A renewed and unprecedented multi-disciplinary research effort from chemists, biochemists, molecular and cell biologists, physiologists, pharmacologists and psychologists is now needed in order to provide grounds to this feeling, and, possibly, to improve our understanding of some basic aspects of mammalian physiology and to exploit this knowledge for the development of novel therapeutic strategies for the treatment and cure of some CNS and peripheral dysfunctions.
The authors wish to thank Prof. Raphael Mechoulam, The Hebrew University of Jerusalem, Israel, for fruitul discussions and for sharing results from work in progress. This article was supported by funds from the Human Frontiers in Science Program Organization (RG 26/95 to VDM) and from the P.O. CNR-MURST ‘Fondi Strutturali 1994-1999’.
1) Devane WA, Dysarz FA, Johnson MR, Melvin LS and Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 34 (1988) 605-613. 2) Matsuda LA, Lolait SJ, Brownstein MJ, Young AC and Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346 (1990) 561-564. 3) Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A and Mechoulam R. Isolation and structure of a brain costituent that binds to the cannabinoid receptor. Science 258 (1992) 1946-1949. 4) Pert CB, Snyder SH. Opiate receptor: demonstration in nervous tissue. Science 179 (1973) 1011-1014. 5) Hughes J, Smith TW, Kosterlitz, HW, Fothergill LA, Morgan BA and Morris HR, Identification of two related pentapeptides from the brain with potent opiate agonist activity.Nature, 258 (1975), 577-579. 6) Mechoulam R. The pharmacohistory of Cannabis sativa. In:Mechoulam R ed. Cannabinoids as Therapeutic agents. Boca Raton: CRC Press (1986) 1-20. 7) Gaoni Y and Mechoulam R. Isolation, structure and partial synthesis of an active constituent of hashish. J Am Chem Soc 86 (1964) 1646-1647. 8) Adams R. Maharihuana. Harvey Lect 37 (1942) 168-197. 9) Mechoulam R ed. Cannabinoids as therapeutic agents. Boca Raton:CRC (1986). 10) Razdan RK. The total synthesis of cannabinoids. In: ApSimon J ed. The total synthesis of natural products. New York: Wiley (1981) 185-262. 11) Razdan RK. Structure-activity relationships in cannabinoids. Pharmacol Rev 32 (1986) 75-149. 12) Martin BR. Cellular effects of cannabinoids. Pharmacol Rev 38 (1986) 45-74. 13) Dewey WL. Cannabinoid pharmacology. Pharmacol Rev 38 (1986) 151-178. 14) Burstein S. Eicosanoids as mediators of cannabinoid action. In: Marijuana/Cannabinoids (Murphy L and Burtke A Eds.), CRC Press, Boca Raton, (1992) 73-91. 15) Martin BR, Compton DR, Thomas BF, Prescott WR, Little PJ, Razdan RK, Johnson MR, Melvin LS, Mechoulam R and Ward SJ. Behavioral, biochemical, and molecular modeling evalutations of cannabinoid analogs. Pharmacol Biochem Behav 40 (1991) 471-478. 16) Mechoulam R, Devane WA and Glaser R. Cannabinoid geometry and biological activity. In: Marijuana/Cannabinoids: Neurobiology and Neurophysiology. Murphy L and Bartke A eds. Boca Raton: CRC Press (1992) 1-33. 17) Howlett AC. Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol 35 (1995) 607-634. 18) Martin BR, Thomas BF and Razdan RK. Structural requirements for cannabinoid receptor probes. In: Pertwee R ed. Cannabinoid Receptors. London: Academic Press (1995) 35-79. 19) Little PJ, Compton DR, Mechoulam R, Martin BR. Stereochemical effects of 11-OH-D8-THC-dimethylheptyl in mice and dogs. Pharmacol Biochem Behav 22 (1989) 661-666. 20) Reggio PH, Seltzman HH, Compton DR, Prescott WR and Martin BR. Investigation of the role of the phenolic hydroxyl in cannabinoid activity. Mol Pharmacol 38 (1990) 40-53. 21) Hollister LE. Structure-activity relationships in man of cannabis constituents, and homologs and metabolites of D9-trtrahydrocannabinol. Pharmacology 11 (1974) 3-11. 22) Lemberger L, Crabtree RE and Rowe HM. 11-Hydroxy-delta-tetrahydro-cannabinol: pharmacology, disposition, and metabolism of a major metabolite in man. Science 177 (1972) 62-63. 23) Compton DR, Johnson MR, Melvin LS and Martin BR. Pharmacological evaluation of a series of bycyclic cannabinoid analogs: classification as cannabinomimetic agents. J Pharmacol Exp Ther 263 (1992) 201-209. 24) Pacheco M, Childers SR, Arnold R, Casiano F and Ward SJ. Amino-alkylindoles: actions on specific G-protein-linked receptors. J Pharmacol Exp Ther 257 (1991) 170-183. 25) Thomas BF, Compton DR, Martin BR and Semus SF. Modeling the cannabinoid receptor: a three-dimensional quantitative structure-activity analysis. Mol Pharmacol 40 (1991) 656-665. 26) Howlett AC. Cannabinoid compounds and signal transduction mechanisms. In: Pertwee R ed. Cannabinoid Receptors. London: Academic Press (1995) 167-204. 27) Mackie K and Hille B. Cannabinoids inhibit N-type calcium current in neuroblastoma-glioma cells. Proc Natl Acad Sci USA 89 (1992) 3825-3829. 28) Gifford AN and Ashby CR. Electrically evoked acetylcholine release from hippocampal slices is inhibited by the cannabinoid receptor agonist, WIN 55,212-2, and is potentiated by the cannabinoid antagonist, SR 141716A. J Pharmacol Exp Ther 277 (1996) 1431-1436. Tripathi HL, Vocci FJ, Brase DA and Dewey WL. Effects of cannabinoids on levels of acetylcholine and choline and on turnover rate of acetylcholine in various regions of the mouse brain. Alcohol Drug Res 7 (1987) 525-532. Maneuf YP, Crossman AR and Brotchie JM. Modulation of GABAergic transmission in the globus pallidus by the synthetic cannabinoid WIN 55,212-2. Synapse 22 (1996) 382-385. Shen MX, Piser TM, Seybold VS and Thayer SA. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosc 16 (1996) 4322-4334. 29) Kaminsky NE, Abood ME, Kessler FK, Martin BR and Schatz AR. Identification of a functionally relevant cannabinoid receptor on mouse spleen cells that is involved in cannabinoid-mediated immune modulation. Mol Pharmacol 42 (1992) 736-742. 30) Lynn AB and Herkenham M. Localization of cannabinoid receptor and nonsaturable high-density cannabinoid binding sites in peripheral tissues of the rat: implications for receptor-mediated immune modulation by cannabinoids. J Pharmacol Exp Ther 268 (1994) 1612-1623. Bouaboula M, Rinaldi M, Carayon P, Carillon C, Delpech B, Shire D, Le Fur G and Casellas P. Cannabinoid-receptor expression in human leukocytes. Eur J Biochem 214 (1993) 173-180. 31) G�rard CM, Mollereau C, Vassart G and Parmentier M. Molecular cloning of a human cannabinoid receptor which is also expressed in testis. Biochem J 279 (1991) 129-134. 32) G�rard CM, Mollereau C, Vassart G and Parmentier M. Nucleotide sequence of a human cannabinoid receptor cDNA. Nucleic Acids Res 18 (1990) 7142-134. 33) Herkenham M. Cannabinoid-receptor localization in the brain and periphery. In: Pertwee R ed. Cannabinoid Receptors. London: Academic Press (1995) 145-166. 34) Matsuda LA and Bonner TI. Molecular biology of the cannabinoid receptor. In: Pertwee R ed. Cannabinoid Receptors. London: Academic Press (1995) 117-143. 35) Munro S, Thomas KL and Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 365 (1993) 61-65. 36) Facci L, Dal Toso R, Romanello S, Buriani A, Skaper SD and Leon A. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc Natl Acad Sci USA 92 (1995) 3376-3380. 37) Felder CC, Veluz JS, Williams HL, Briley EM and Matsuda LA. Cannabinoid agonists stimulate both receptor- and non-receptor-mediated signal transduction pathways in cells transfected with and expressing cannabinoid receptor clones. Mol Pharmacol 42 (1992) 838-845. 38) Deadwyler SA, Hampson RE, Bennett BA, Edwards TA, Mu J, Cannabinoids modulate potassium current in cultured hippocampal neurons. Recept Channels 1 (1993) 121-134. 39) Mackie K, Lai Y, Westenbroek R and Mitchell R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci 15 (1995) 6552-6561. 41) Bouaboula M, Poinotchazel C, MarchandJ, Canat X, Bourrie B, Rinaldi-Carmona M, Calandra B, Le Fur G and Casellas P. Signalling pathway associated with stimulation of CB2 peripheral cannabinoid receptor. Involvement of both mitogen-activated protein kinase and induction of Krox-24 expression. Eur J Biochem 237 (1996) 704-711. 42) Bouaboula M, Poinot-Chazel C, Bourrie B, Canat X, Calandra B, Rinaldi-Carmona M, Le Fur G and Casellas P. Activation of mitogen-activated protein kinase by stimulation of the central cannabinoid receptor CB1. Biochem J 312 (1995) 637-641. 43) Rinaldi-Carmona M, Barth F, H�aulme M, Shire D, Calandra B, Congy C, Martinez S, Maurani J, Neliat G, Caput D, Ferrara P, Soubri� P, Breliere JC and Le Fur G. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett 350 (1994) 240-244. 44) Bayewitch M, Rhee RH, Avidor-Reiss T, Breuer A, Mechoulam R and Vogel Z. (-)-Delta-(9)-Tetrahydro-cannabinol antagonizes the peripheral cannabinoid receptor-mediated inhibition of adenylyl cyclase. J Biol Chem 271 (1996) 9902-9905. 45) Showalter VM, Compton DR, Martin BR and Abood ME. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2). Identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther 278 (1996) 989-999. 46) Gallant M, Dufresne C, Gareau Y, Guay D, Leblanc Y, Prasit P, Rochette C, Sawyer N, Slipetz DM, Tremblay N, Metters KM and Labelle M. New class of potent ligands for the human peripheral cannabinoid receptor. Biorg Med Chem Lett 6 (1996) 2263-2268. 47) Shire D, Calandra B, Delpech M, Dumont X, Kaghad M, Le Fur G, Caput D and Ferrara P. Structural features of the central cannabinoid receptor involved in the binding of the specific CB1 antagonist SR141716A. J. Biol. Chem. 271 (1996) 6941-6946. 48) Shire D, Carillon C, Kaghad M, Calandra B, Rinaldi-Carmona M, Le Fur G, Caput D and Ferrara P. An amino-terminal variant of the central cannabinoid receptor resulting from alternative splicing. J Biol Chem 270 (1995) 3726-3731. 49) Evans DM, Lake JT, Johnson MR and Howlett AC. Endogenous cannabinoid receptor binding activity released from rat brain slices by depolarization. J Pharmacol Exper Ther 268 (1994) 1271-1277. 50) Pertwee R. Pharmacological, physiological and clinical implications of the discovery of cannabinoid receptors: an overview. In: Pertwee R ed. Cannabinoid Receptors. London: Academic Press (1995) 1-34. 51) Fride E and Mechoulam R. Pharmacological activity of the cannabinoid agonist anandamide, a brain constituent. Eur J Pharmacol 231 (1993) 313-314. 52) Crawley JN, Corwin RL, Robinson JK, Felder CC, Devane WA and Axelrod J. Anandamide, an endogenous ligand of the cannabinoid receptor, induces hypomotility and hypothermia in vivo in rodents. Pharmacol Biochem Behav 46 (1993) 967-972. 53) Smith PB, Compton DR, Welch SP, Razdan RK, Mechoulam R and Martin BR. The pharmacological activity of anandamide, a putative endogenous cannabinoid, in mice. J Pharmacol Exp Ther 270 (1994) 219-227. 54) Felder CC, Briley EM, Axelrod J, Simpson JT, Mackie K and Devane WA. Anandamide, an endogenous cannabinomimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proc Natl Acad Sci USA 90 (1993) 7656-7660. 55) Vogel Z, Barg J, Levy R, Saya D, Heldman E and Mechoulam R. Anandamide, a brain endogenous compound, interacts specifically with cannabinoid receptors and inhibits adenylate cyclase. J Neurochem 61 (1993) 352-355. 56) Mackie K, Devane WA and Hille B. Anandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Mol Pharmacol 44 (1993) 498-503. 57) Van der Kloot W. Anandamide, a naturally occurring agonist of the cannabinoid receptor, blocks adenylate cyclase at the frog neuromuscolar junction. Brain Res 649 (1994) 181-184. 58) Mallet PE and Beninger RJ. The endogenous cannabinoid receptor agonist anandamide impairs memory in rats. Behav Pharmacol 7 (1996) 276-284. 59) Lichtman AH, Dimen KR and Martin BR. Systemic or intrahippocampal cannabinoid administration impairs spatial memory in rats. Psichopharmacology 119 (1995) 282-290. 60) Romero J, Garcia L, Cebeira M, Zadrozny D, Fernandez-Ruiz JJ and Ramos JA. The endogenous cannabinoid receptor ligand, anandamide, inhibits the motor behavior: Role of nigrostriatal dopaminergic neurons. Life Sci 56 (1995) 2033-2040. 61) Terranova JP, Michaud JC, Lefur G and Soubrie P. Inhibition of long-term potentiaton in rat hippocampal slices by anandamide and WIN55212-2: reversal by SR141716A, a selective antagonist of CB1 cannabinoid receptors. Naunyn Schmiedeberg's Arch Pharmacol 352 (1995) 576-579. 62) Souilhac J, Poncelet M, Rinaldi-CarmonaM, Le-Fur G and Soubrie P. Intrastrial injection of cannabinoid receptor agonists induced turning behavior in mice. Pharmacol Biochem Behav 51 (1995) 3-7. 63) Pate DW, Jarvinen K, Urtti A, Jarho P and Jarvinen T. Ophthalmic arachidonylethanolamide decreases intraocular pressure in normotensive rabbits. Current Eye Res 14 (1995) 791-797. 64) Varga K, Lake K, Martin BR and Kunos G. Novel antagonist implicates the CB1 cannabinoid receptor in the hypothensive action of anandamide. Eur J Pharmacol 278 (1995) 279-283. 65) Weidenfeld J, Feldman S and Mechoulam R. Effect of the brain costituent anandamide, a cannabinoid receptor agonist, on the hypothalamo-pituitary-adrenal axis in the rat. Neuroendocrinology 59 (1994) 110-112. 66) Wenger T, Toth BE and Martin BR. Effects of anandamide (endogen cannabinoid) on anterior pituitary hormone secretion in adult ovariectomized rats. Life Sci 56 (1995) 2057-2063. 67) Giannikou P, Yiannakakis N, Frangkakis G, Probonas K and Wenger T. Anandamide (endogen cannabinoid) decreases serum prolactin level in pregnant rat. Neuroendocinol Lett 17 (1995) 281-287. 68) Schlicker E, Timm J and Gother M. Cannabinoid receptor-mediated inhibition of dopamine release in the retina. Naunyn Schmiedeberg's Arch Pharmacol 354 (1996) 791-795. 69) Ishac EJN, Jiang L, Lake KD, Varga K, Abood ME and Kunos G. Inhibition of exocytotic noradrenaline release by presynaptic cannabinoid CB1 receptors on peripheral sympathetic nerves. Br J Pharmacol 118 (1996) 2023-2028. 70) Wickens AP and Pertwee RG. D9-Tetrahyrocannabinol and anandamide enhance the ability of muscimol to induce catalepsy in the globus pallidus of rats. Eur J Pharmacol 250 (1993) 205-208. 71) Romero J, Garcia-Palomero E, Fernandez-Ruiz JJ and Ramos JA. Involvement of GABA(B) receptors in the motor inhibition produced by agonists of brain cannabinoid receptors. Behav Pharmacol 7 (1996) 299-302. 72) Cabral GA, Toney DM, Fischer-Stenger K, Harrison MP and Marciano-Cabral F. Ananadamide inhibits macrophage-mediated killing of tumor necrosis factor-sensitive cells. Life Sci 56 (1995) 2065-2072. 73) Schwarz H, Blanco FJ and Lotz M. Ananadamide, an endogenous cannabinoid receptor agonist inhibits lymphocyte proliferation and induces apoptosis. J Neuroimmunol 55 (1994) 107-115. 74) Schuel H, Goldstein E, Mechoulam R, Zimmerman AM and Zimmerman S. Anandamide (arachidoylethanolamide), a brain cannabinoid receptor agonist, reduces sperm fertilizing capacity in sea urchins by inhibiting the acrosome reaction.Proc Natl Acad Sci USA 91 (1994) 7678-7682. 75) Das SK, Paria BC, Chakraborty I and Dey SK. Cannabinoid ligand-receptor signaling in the mouse uterus. Proc Natl Acad Sci USA 92 (1995) 4332-4336. 76) Paria BC, Das SK and Dey SK. The preimplantation mouse embryo is a target for cannabinoid ligand-receptor signalling. Proc Natl Acad Sci USA 92 (1995) 9460-9464. 77) Pertwee RG RG, Stevenson LA and Griffin G. Cross-tolerance between delta-9-tetrahydrocannabinol and the cannabimimetic agents, CP 55,940, WIN 55,212-2, and anandamide. Br J Pharmacol 110 (1993)1483-1490. 78) Fride E. Anandamides: tolerance and cross-tolerance to delta(9)-tetrahydrocannabinol. Brain Res 697 (1995) 83-90. 79) Welch SP, Dunlow LD, Patrick GS and Razdan RK. Characterization of anandamide- and fluoroanandamide-induced antinociception and cross-tolerance to delta-9-THC after intrathecal administration to mice: blockade of delta-9-THC induced antinociception. J Pharmacol Exp Ther 273 (1995) 1235-1244. 80) Romero J, Garcia L, Fernandez-Ruiz JJ, Cebeira M and Ramos JA. Changes in rat brain cannabinoid binding sites after acute or chronic exposure to their endogenous agonist, anandamide or to delta-9-tetrahydrocannabinol. Pharmacol Biochem Behav 51 (1995) 731-737. 81) Fride E, Barg J, Levy R, Low doses of anandamides inhibit pharmacological effects of delta-9-tetra-hydrocannabinol. J Pharmacol Exp Ther 272 (1995) 699-707. 82) Federman AD, Conklin BR, Schrader KA, Reed RR and Bourne HR. Hormonal stimulation of adenylyl cyclase through Gi-protein bg subunits. Nature 356 (1992) 159-161. 83) Koga D, Santa T, Hagiwara K, Imai K, Takizawa H, Nagano T, Hirobe M, Ogawa M, Sato T, Inoue K and Kudo I. High performance liquid chromatography and fluorimetric detection of arachidonylethanolamide (anandamide) and its analogues, derivatized with 4-(N-chloroformylmethyl-N-methyl)amino-7-N-,N-dimethylaminosulphonyl-2,1,3-benzoxadiazole (DBD-COCl). Biomed Chromatogr 9 (1995) 56-57. 84) Sugiura T, Kondo S, Sukagawa A, Tonegawa T, Nakane S, Yamashita A and Waku K. Enzymatic synthesis of anandamide, an endogenous cannabinoid receptor ligand, through N-acylphosphatidylethanolamine pathway in testis: involvement of Ca2+-dependent transacylase and phosphodiesterase activities. Biochem Biophys Res Comm 218 (1996) 113-117. 85) Fontana A, Di Marzo V, Cadas H and Piomelli D. Analysis of ananadamide, an endogenous cannabinoid substance, and other natural N-acylethanolamines. Prostag Leuk Ess Fatty Acids 53 (1995) 301-308. 86) Schmid PC, Krebsbach RJ, Perry SR, Dettmer TM, Maassen JL and Schmid HHO. Occurrence and postmortem generation of ananadamide and other long-chain N-acylethanolamines in mammalian brain. FEBS Lett 375 (1995) 117-120. 87) Kempe K, Hsu FF, Bohrer A and Turk J. Isotope dilution mass spectrometric measurements indicate that arachidonyletanolamide, the proposed endogenous ligand of the cannabinoid receptor, accumulates in rat brain tissue post mortem but is contained at low levels in or absent from fresh tissue. J Biol Chem 271 (1996) 17287-17295. 88) Felder CC, Nielsen A, Briley EM, Palkovitis M, Priller J, Axelrod J, Nguyen DN, Richardson JM, Riggin RM, Koppel GA, Paul SM and Becker GW. Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS Lett 393 (1996) 231-235. 89) Sugiura T, Kondo S, Sukagawa A, Tonegawa T, Nakane S, Yamashita A, Ishima Y and Waku K. Transacylase-mediated and phosphodiesterase-mediated synthesis of N-arachidonoylethanolamine, an endogenous cannabinoid-receptor ligand, in rat brain microsomes. Comparison with the synthesis from free arachidonic acid and ethanolamine. Eur J Biochem 240 (1996) 53-62. 90) Childers SR, Sexton T and Roy MB. Effects of anandamide on cannabinoid receptors in rat brain membranes. Biochem Pharmacol 47 (1994) 711-715. Pertwee RG, Fernando SR, Griffin G, Abadji V and Makriyannis A. Effect of phenylmethylsulphonyl fluoride on the potency of anandamide as an inhibitor of electrically evoked contractions in two isolated tissue preparations. Eur J Pharmacol 272 (1995) 73-78. 91) Hampson AJ, Hill WA, Zan-Phillips M, Makriyannis A, Leung E, Eglen RM and Bornheim LM. Anandamide hydroxylation by brain lipoxygenase: metabolite structures and potencies at the cannabinoid receptor. Biochem Biophys Acta 1259 (1995) 173-179. 92) Bornheim LM, Kim KY, Chen B and Correia MA. The effect of cannabidiol on mouse hepatic microsomal cytochrome P-450-dependent anandamide metabolism. Biochem Biophys Res Comm 197 (1993) 740-746. 93) Ueda N, Yamamoto K, Yamamoto S, Tokunaga T, Shirakawa E, Shinaki H, Ogawa M, Sato T, Kudo I, Inoue K, Takizawa H, Nagano T, Hirobe M and Saito H. Lipoxygenase-catalyzed oxygenation of arachidonylethanolamide, a cannabinoid receptor agonist. Biochem Biophys Acta 1254 (1995) 127-134. 94) Pinto JC, Potie F, Rice KC, Boring D, Johnson MR, Evans DM, Wilken GH, Cantrell CH and Howlett. Cannabinoid receptor binding and agonist activity of amides and esters of arachidonic acid. Mol Pharmacol 46 (1994) 516-522. 95) Adams IB, Ryan W, Singer M, Thomas BF, Compton DR, Razdan RK and Martin BR. Evaluation of cannabinoid receptor binding and in vivo activities for anandamide analogs. J Pharmacol Exp Ther 273 (1995) 1172-1181. 96) Edgemond WS, Campbell WB and Hillard CJ. The binding of novel phenolic derivatives of anandamide to brain cannabinoid receptors. Prostag Leuk Ess Fatty Acids 52 (1995) 83-86. 97) Priller J, Briley EM, Mansouri J, Devane WA, Mackie K and Felder CC. Mead ethanolamide, a novel eicosanoid, is a agonist for the central (CB1) and peripheral (CB2) cannabinoid receptors. Mol Pharmacol 48 (1995) 288-292. 98) Abadji V, Lin S, Taha G, Griffin G, Stevenson LA, Pertwee RG and Makriyannis A. (R)-methanandamide: a chiral novel anandamide possessing higher potency and metabolic stability. J Med Chem 37 (1994) 1889-1893. 99) Di Marzo V, De Petrocellis L, Bisogno T and Maurelli S. Pharmacology and physiology of the endogenous cannabimimetic mediator anandamide. J Drug Dev Clin Pract 7 (1995) 199-219. 100) Thomas BF, Adams IB, Mascarella SW, Martin BR and Razdan RK. Structure-activity analysis of anandamide analogs: relationship to a cannabinoid pharmacophore. J Med Chem 39 (1996) 471-479. 101) Hanus L, Gopher A, Almog S and Mechoulam R. Two new unsatured fatty acid ethanolamides in brain that bind to cannabinoid receptors. J Med Chem 36 (1993) 3032-3034. 102) Pertwee R, Griffin G, Hanus L and Mechoulam R. Effects of two endogenous fatty acid ethanolamides on mouse vasa deferentia. Eur J Pharmacol 259 (1994) 115-120. 103) Barg J, Fride E, Hanus L, Levy R, Matus-Leibovitch N, Heldman E, Bayewitch M, Mechoulam R and Vogel Z. Cannabinomimetic behavioral effects of and adenylate cyclase inhibition by two new endogenous anandamides. Eur J Pharmacol 287 (1995) 145-152. 104) Sheskim T, Hanus L, Slager J, Vogel Z and Mechoulam R. Structural requirements for binding of anandamide-type compounds to the brain cannabinoid receptor. J Med Chem 40 (1997) 659-667. 105) Khanolkar AD, Abadji V, Lin S, Hill WA, Taha G, Abouzid K, Meng Z, Fan P, Makriyannis A. Head group analogs of arachidonylethanolamide, the endogenous cannabinoid ligand. J Med Chem 39 (1996) 4515-4519. 106) Bayewitch M, Avidor-Reiss T, Levy R, Barg J, Mechoulam R and Vogel Z. The peripheral cannabinoid receptor: adenylate cyclase inhibition and G protein coupling. FEBS Lett 375 (1995) 143-147. 107) Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J and Vogel Z. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50 (1995) 83-90. 108) Lee M, Yang KH and Kaminski NE. Effects of putative cannabinoid receptor ligands, anandamide and 2-arachidonoyl-glycerol on immune function in B6C3F1 mouse splenocytes. J Pharmacol Exp Ther 275 (1995) 529-536. 109) Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A and Waku K. 2-Arachidonoyl-glycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Comm 215 (1995) 89-97. 110) Shohami E, Weidenfeld J, Ovadia H, Vogel Z, Hanus L, Fride E, Breuer A, Ben-Shabat S, Sheskin T and Mechoulam R. Endogenous and synthetic cannabinoids: recent advances. CNS Drug Rev 4 (1997) 429-451. 111) Schmid HHO, Schmid PC and Natarajan V. N-Acylated glicerophospholipids and their derivatives. Prog Lipid Res 29 (1990) 1-43. 112) Mazzari S, Canella R, Petrelli L, Marcolongo G and Leon A. N-(2-hydroxyethyl)hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. Eur. J. Pharmacol 300, (1996) 227-236. 113) Bisogno T, Maurelli S, Melck D, De Petrocellis L and Di Marzo V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J Biol Chem 272 (1997) 3315-3323. 114) Skaper SD, Buriani A, Dal Toso R, Petrelli L, Romanello S, Facci L and Leon A. The ALIAmide palmitoylethanolamide and cannabinoids, but not anandamide, are protective in a delayed postglutamate paradigm of exocitotic death in cerebellar granule neurons.Proc Natl Acad Sci USA 93 (1995) 3984-3989. 115) Feigenbaum JJ, Bergmann F, Richmond SA, Mechoulam R, Nadler V, Kloog Y and Sokolowsky M. Non-psychotropic cannabinoid acts as a functional N-methyl-D-aspartate receptor blocker. Proc Natl Acad Sci USA 86 (1989) 9584-9587. 116) Cravatt BF, Prospero-Garcia O, Siuzdak G, Gilula NB, Henriksen SJ, Boger DL and Lerner RA. Chemical characterization of a family of brain-lipids that induce sleep. Science 268 (1995) 1506-1509. 117) Mechoulam R, Fride E, Hanus L, Sheskin T, Bisogno T, Di Marzo V, Bayewitch M, Vogel Z, nandamide may mediate sleep induction', Nature, 389, 25-26. 118) Santucci V, Storme JJ, Soubrie P and Le Fur G. Arousal-enhancing properties of the CB1 cannabinoid receptor antagonist SR 141716A in rats as assessed by electroencephalographic spectral and sleep-waking cycle analysis. Life Sci 58 (1996) 103-110. 119) Venance L, Piomelli D, Glowinski J and Giaume C. Inhibition by anandamide of gap junctions and intercellular calcium signalling in striatal astrocytes. Nature 376 (1995) 590-594. 120) Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA and Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty acid amides. Nature 384 (1996) 83-87. 121) Langstein J, Hofstadter F and Schwartz H. Cis-9,10-octadecenoamide, an endogenous sleep-inducing CNS compound, inhibits lymphocytes proliferation. Res Immunol 147 (1996) 389-396. 122) Boring DL, Berglund BA and Howlett AC. Cerebrodiene, arachidonyl-ethanolamide, and hybrid structures: potential for interaction with cannabinoid receptors. Prostag Leuk Ess Fatty Acids 55 (1996) 207-210. 123) Maurelli S, Bisogno T, De Petrocellis L, Di Luccia A, Marino G and Di Marzo V. Two novel class of neuroactive fatty acid amides are substates for mouse neuroblastoma anandamide amidohydrolase. FEBS Lett 377 (1995) 82-86. 124) Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC and Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372 (1994) 686-691. 125) Schmid PC, Reddy PV, Natarajan V and Schmid HHO. Metabolism of N-acylethanolamine phospholipids by a mammalian phosphodiesterase of the phospholipase D type. J. Biol. Chem. 258 (1983) 9302-9306. 126) Kruszka KK and Gross RW. The ATP- and CoA-independent synthesis of arachidonoylethanolamide. J. Biol. Chem. 269 (1994) 14345-14348. 127) Devane WA and Axelrod J. Enzymatic synthesis of anandamide, an endogenous ligand for the cannabinoid receptor, by brain membranes. Proc Natl Acad Sci USA 91 (1994) 6698-6701. 128) Ueda N, Kurahashi Y, Yamamoto S and Tokunaga T. Partial purification and characterization of the porcine brain enzyme hydrolazing and synthesizing anandamide. J Biol Chem 270 (1995) 23823-23827. 129) Cadas H, Gaillet S, Beltramo M, Venance L and Piomelli D. Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J Neurosci 16 (1996) 3934-3942. Cadas H , di Tomaso E and Piomelli D, Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl-phosphatidylethanolamine, in rat brain. J Neurosci 17 (1997) 1226-1242. 130) Natarajan V, Schmid PC, Reddy PV, Zuzarte-Augustin M and Schmid HHO. Biosynthesis of N-acylethanolamine phospholipids by dog brain preparation. J Neurochem 41 (1983) 1303-1312. 131) Burstein SH and Hunter SA. Stimulation of anandamide biosynthesis in N18TG2 neuroblastoma cells by D9-tethrahydrocannabinol (THC). Biochem Pharmacol 49 (1995) 855-858. Di Marzo V, De Petrocellis L, Sepe N and Buono A. Biosynthesis of anandamide and related acylethanolamides in mouse J774 macrophages and N18 neuroblastoma cells. Biochem J 316 (1996) 977-984. 132) Bisogno T, Ventriglia
M, Mosca M, Milone A, Cimino G and Di Marzo V. Occurrence and metabolism of anandamide and
related acylethanolamides in ovaries of the sea urchin Paracentrotus lividus. Biochim.
Biophys. Acta, 1345 (1997) 338-348. 133) Paria BC, Deutsch DD and Dey SK. The uterus is a potential site for ananadamide synthesis and hydrolysis: differential profiles of ananadamide synthase and hydrolase activities in the mouse uterus during the periimplantation period. Mol Repr Develop 45 (1996) 183-192. 134) Di Marzo V and Fontana A. Anandamide, an endogenous cannabinomimetic eicosanoid: 'killing two birds with one stone'. Prostag Leuk Ess Fatty Acids 53 (1995) 1-11. Schmid HHO, Schmid PC and Natarajan V. The N-acylation-phosphodiesterase pathway and cell signalling. Chem Phys Lipids 80 (1996) 133-142. 135) Bisogno T, Sepe N, Melck D, Maurelli S, De Petrocellis L and Di Marzo V. Biosynthesis, release and degradation of the novel endogenous cannabinomimetic metabolite 2-arachidonoyl-glycerol in mouse neuroblastoma cells. Biochem J 322 (1997) 671-677. 136) Di Marzo V, De Petrocellis L, Sugiura T and Waku K. Potential biosynthetic connections between the two cannabimimetic eicosanoids, anandamide and 2-arachidonoyl-glycerol, in mouse neuroblastoma cells. Biochem Biophys Res Comm 227 (1996) 281-288. 137) Deutsch DG and Chin SA. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol 46 (1993) 791-796. 138) Desarnaud F, Cadas H and Piomelli D. Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J Biol Chem 270 (1995) 6030-6035. 139) Hillard CJ, Wilkison DM, Edgemond WS and Campbell WB. Characterization of the kinetics and distribution of N-arachidonylethanolamine (anandamide) hydrolysis by rat brain. Biochim Biophys Acta 1257 (1995) 249-256. 140) Watanabe K, Kayano Y, Matsunaga T, Yamamoto I and Yoshimura H. Inhibition of anandamide amidase activity in mouse brain microsomes by cannabinoids. Biol Pharmaceut Bull 19 (1997) 1109-1111. 141) Koutek B, Prestwich GD, Howlett AC, Chin SA, Salehani D, Akhavan N and Deutsch DG. Inhibitors of arachidonyl ethanolamide hydrolysis. J Biol Chem 269 (1994) 22937-22940. 142) Cravatt BF, Lerner RA and Boger DL. Structure determination of an endogenous sleep-inducing lipid, cis-9-octadecenamide (oleamide): a synthetic approach to the chemical analysis of traces quantities of a natural product. J Am Chem Soc 118 (1996) 580-590. 143) Patterson JE, Ollman IR, Cravatt BF, Boger DL, Wong G-H and Lerner RA. Inhibition of oleamide hydrolase catalyzed hydrolysis of the endogenous sleep-inducing lipid cis-9-octadecenamide. J Am Chem Soc 118 (1996) 5938-5945. 144) Matsuda S, Kanemitsu N,Nakamura A, Mimura Y, Ueda N, Kurahashi Y and Yamamoto S. Metabolism of anandamide, an endogenous cannabinoid receptor ligand, in porcine ocular tissues. Exp Eye Res 64 (1997) 707-711 145) De Petrocellis L, Melck D, Ueda N, Maurelli S, Kurahashi Y, Yamamoto S, Marino G and Di Marzo V. Novel inhibitors of brain, neuronal and basophilic anandamide aminohydrolase. Biochem Biophys Res Comm 231(1997) 82-88. Deutsch DG, Omeir R, Arreaza G, Salehani D, Prestwich GD, Huang Z and Howlett A. Methylarachidonyl-fluorophosphonate: a potent irreversible inhibitor of anandamide amidase. Biochem Pharmacol 53 (1997) 255-260. 146) Lang WS, Qin C, Hill WAG, Lin SY, Khanolkar AD and Makriyannis A. High performance liquid chromatography determination of anandamide amidase activity in rat brain microsomes. Anal Biochem 238 (1996) 40-45. 147) Wise ML, Hamberg M and Gerwick WH. Biosynthesis of conjugated triene-containing fatty acids by a novel isomerase from the red marine alga Ptilota filicina. Biochemistry 33 (1994) 15223-15232. 148) Wise ML, Soderstrom K, Murray TF and Gerwick WH. Synthesis and cannabinoid receptor binding activity of conjugated triene anandamide, a novel eicosanoid. Experientia 52 (1996) 88-92. 149) Ben-Shabat S, Fride E, Vogel Z, Bisogno T, De Petrocellis L, Di Marzo V and Mechoulam R, manuscript in preparation. 150) Hillard CJ, Edgemond WS, Jarrahian A and Campbell WB. 'Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion' J. Neurochem 69 (1997) 631-638 151) Derkinderen P, Toutant M, Burgaya F, Le Bert M, Siciliano JC, de Franciscis V, Gelman M and Girault J-A. Regulation of a neuronal form of focal adhesion kinase by anandamide. Science 273 (1996) 1719-1722. 152) Pugh G, Smith PB, Dombrowski DS and Welch SP. The role of endogenous opioids in enhancing the antinociception produced by the combination of Delta(9)-tetrahydrocannabinol and morphine in the spinal cord. J Pharmacol Exp Ther 279 (1996) 608-616. 153) Di Marzo V, De Petrocellis L, Bisogno T, and Maurelli S. The endogenous cannabimimetic eicosanoid, anandamide, induces arachidonate release in J774 mouse macrophages. In: Eicosanoids and other bioactive lipids in cancer, inflammation and radiation injury (Honn KV, Marnett LJ, Wong P Y-K, Jones R and Nigam S Eds.), Plenum Press, Adv Exp Med Biol 407 (1997) 341-346 154) Payan DG, Levine JD and Goetzl EJ. Modulation of immunity and hypersensitivity by sensory neuropeptides. J Immunol 132 (1984) 1601-1604. 155) Richardson JD, Garry MG, Jackson DL, Aanonsen LM and Hargreaves KJ. Anandamide suppresses capsaicin-evoked iSP and iCGRP release in the dorsal horn of rat spinal cord. Soc Neurosci Abs 20 (1994) 128. 156) Stefano GB, Liu Y and Goligorsky MS. Cannabinoir receptors are coupled to nitric oxide release in invertebrate immunocytes, microglia, and human monocytes. J Biol Chem 271 (1996) 19238-19242. 157) Coffey RG, Yamamoto Y, Snella E and Pross S. Tetrahydrocannabinol inhibition of macrophage nitric oxide production. Biochem Pharmacol 52 (1996) 743-751. 158) Pertwee RG, Fernando SR, Nash JE and Coutts AA. Further evidence for the presence of cannabinoid CB1 receptors in guinea-pig small intestine. Br J Pharmacol 118 (1996) 2199-2205. 159) Pertwee RG and Fernando SR. Evidence for the presence of cannabinoid CB1 receptors in mouse urinary bladder. Br J Pharmacol 118 (1996) 2053-2058. 160) Varga K, Lake K, Martin BR and Kunos G. Novel antagonist implicates the CB1 cannabinoid receptor in the hypothensive action of anandamide. Eur J Pharmacol 278 (1995) 279-283. 161) Varga K, Lake K, Huangfu DH, Guyenet PG and Kunos G. Mechanism of hypothensive action of anandamide in anesthetized rats. Hypertension 28 (1996) 682-686. 162) Pertwee RG, Joeadigwe G and Hawksworth GM. Further evidence for the presence of cannabinoid CB1 receptors in mouse vas deferens. Eur J Pharmacol 296 (1996) 169-172. 163) Chang MC, Berkery D, Schuel R, Laychock SG, Zimmerman AM, Zimmerman S and Schuel H. Evidence for a cannabinoid receptor in sea urchin sperm and its role in blockade of the acrosome reaction. Mol Reprod Develop 36 (1993) 507-516. 164) Schuel H, Berkery D, Schuel R, Chang MC, Zimmerman AM and Zimmerman S. Reduction of the fertilizing capacity of sea urchin sperm by cannabinoids derived from marihuana. I. Inhibition of the acrosome reaction induced by egg jelly. Mol Reprod Develop 29 (1991) 51-59. 165) Fride E and Mechoulam R. Developmental aspects of anandamide: ontogeny of response and prenatal exposure. Psychoneuroendocrinology 21 (1996) 157-172. 166) Song ZH and Bonner TI. A lysine residue of the cannabinoid receptor is critical for the receptor recognition by several agonists but not WIN55212-2. Mol Pharmacol 49 (1996) 891-896. 167) Sugiura T, Kodaka T, Kondo S, Tonegawa T, Nakane S, Kishimoto S, Yamashita A and Waku K. 2-arachidonoyl-glycerol, a putative endogenous cannabinoid receptor ligand, induces rapid, transient elevation of intracellular free Ca2+ in neuroblastoma X glioma hybrid NG108-15 cells. Biochem Biophys Res Commun 229 (1996) 58-64. 168) Sugiura T, Kondo S, Kodaka T, Tonegawa T, Nakane S, Yamashita A, Ishima Y and Waku K. Enzymatic synthesis of oleamide (cis-9,10-octadecenoamide), an endogenous sleep-inducing lipid, by rat brain microsomes. Biochem Mol Biol Int 40 (1996) 931-938. 169) Romero J, Garcia-Palomero E, Berrendero F, Garcia-Gil L, Hernandez ML, Ramos JA and Fernandez-Ruiz JJ. A typical location of cannabinoid receptors in white matter areas during rat brain development. Synapse 26 (1997) 317-323. 170) De Petrocellis L, Orlando P and Di Marzo V. Anandamide, an endogenous cannabinomimetic substance, modulates rat brain protein kinase C in vitro. Biochem Mol Biol Int 36 (1995) 1127-1133. 171) Randall MD, Alexander SPH, Bennett T, Boyd EA, Fry JR, Gardiner SM, Kemp PA, McCulloch AI, and Kendall DA. An endogenous cannabinoid as an endothelium-derived vasorelaxant. Biochem Biophys Res Comm 229 (1996) 114-120. 172) Devane WA. New dawn of cannabinoid pharmacology. Trends Pharmacol Sci 15 (1994) 40-41. |